Danon disease is a rare X-linked dominant disorder caused by mutations in the lysosome-associated membrane protein 2 gene (LAMP2) [1, 2]. Reports on the phenotype of Danon disease are largely focused on severe phenotype of young male patients, who present with the classical triad of massive left ventricular hypertrophy often accompanied by Wolff-Parkinson-White syndrome (WPW) on electrocardiogram, peripheral myopathy and mental retardation [1, 3, 4]. In the present study, we describe variability in clinical expression, namely severe heart failure due to hypertrophic cardiomyopathy leading to heart transplantation at the age of 18 years in the female proband and asymptomatic course without left ventricular hypertrophy or systolic dysfunction in the proband’s mother, both of them carriers of a novel truncating variant in the LAMP2 gene.
The 18-year-old woman with hypertrophic cardiomyopathy diagnosed at 12 years, with complex ventricular arrhythmias leading to implantable cardioverter defibrillator (ICD) treatment in primary prevention was referred to the Institute of Cardiology, Warsaw, due to extreme fatigue, shortness of breath during small activity (30 m walk distance), progressive dyspnea while in a lying position, and periodic blood pressure drops to 60/40 mm Hg as a result of severe heart failure. She had no intellectual impairment. Her father died at the age of 36 years due to an extracardiac cause, and her mother was asymptomatic with normal noninvasive cardiological examinations.
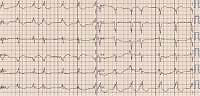
Physically her heart rate was regular, 60 beats per minute, blood pressure was low, 85/60 mm Hg, the stomach was bloated and sensitive with no acute abdominal signs. Cardiac examination revealed no murmur and lung fields were bilaterally clear in auscultation. The liver was enlarged. No peripheral edema was noted. The proband’s standard 12-lead ECG showed sinus rhythm 72 bpm, normal PR interval 124 ms, left bundle branch block with fragmented QRS of 194 ms duration (Figure 1). Transthoracic echocardiography revealed normal left ventricular size with moderate symmetric wall thickening (max. 18.2 mm) without left ventricular outflow tract obstruction and discrete granular sparkling appearance of the interventricular septum (Figure 2). Left ventricular systolic function was severely decreased (LVEF 25%) with contraction asynchrony (Figure 3 A) and reduced left ventricular outflow tract velocity-time integral (11.7 cm). Doppler-derived LV diastolic filling demonstrated a restrictive LV filling pattern (Figure 3 B) and elevated left ventricular filling pressure with E/e′ 18.2 (Figure 3 C). Mitral regurgitation was recorded in systole with a low systolic dP/dt ratio of 241 mm Hg/s but also it was in diastole and concerned decreased left ventricular systolic function and high filling pressure. The left atrium was only slightly widened, 44 mm. Furthermore, noncompaction of the right ventricular myocardium (Figure 3 D) with decreased RV systolic function was noted (TAPSE 10 mm, S′ 5.8 cm/s). The pulmonary artery was widened (3.5 cm) with reduced maximal flow velocity (33 cm/s). Atrioventricular valves were normal. She was diagnosed with hypertrophic cardiomyopathy and right ventricular noncompaction in the phase of severe heart failure with the suggestion of storage disease. Cardiac catheterization revealed normal pulmonary systolic pressure (26 mm Hg) and slightly elevated capillary wedge pressure (15/19/14 mm Hg). Endomyocardial biopsy showed hypertrophic cardiomyocytes with abundant eosinophilic cytoplasm, severe interstitial fibrosis and the presence of vacuoles containing glycogen (Figures 4 A–D). Laboratory tests showed elevated serum transaminases, Nt-proBNP (13981.00 pg/ml) and high-sensitivity troponin T (34.1 ng/ml) concentration and normal CK values (62 U/l). Due to progressive symptoms of heart failure, she was treated with a biventricular assist device for mechanical circulatory support for one month and subsequently received a heart transplant.
Figure 1
Standard 12-lead ECG shows sinus rhythm 72 bpm. Atrial premature complexes. Normal PR interval 124 ms. Left bundle branch block with QRS duration 194 ms, fragmented QRS complex. Prolonged QT duration 503 ms
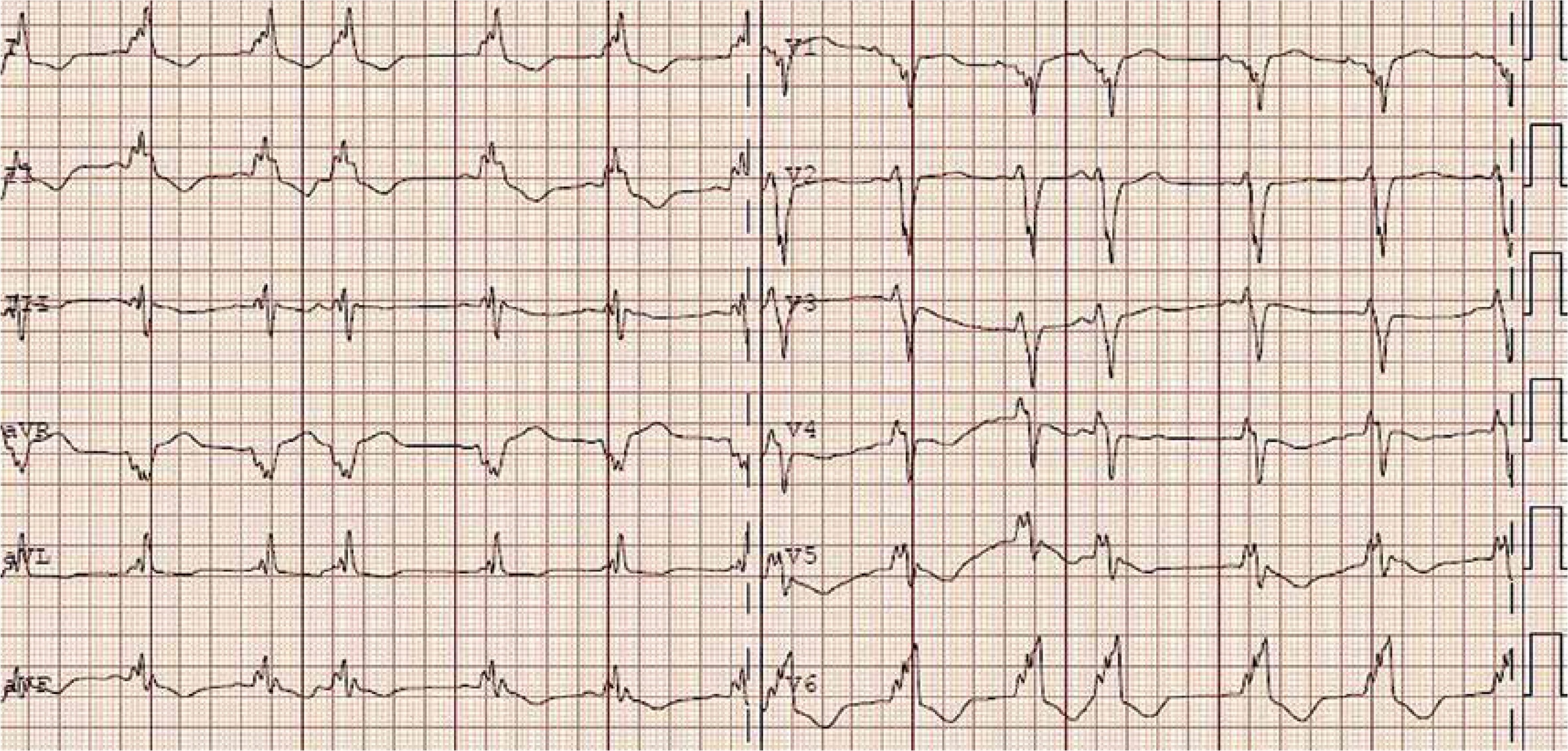
Figure 2
Echocardiogram – parasternal long axis view. Concentric symmetric left ventricle wall thickening with discrete granular sparkling appearance
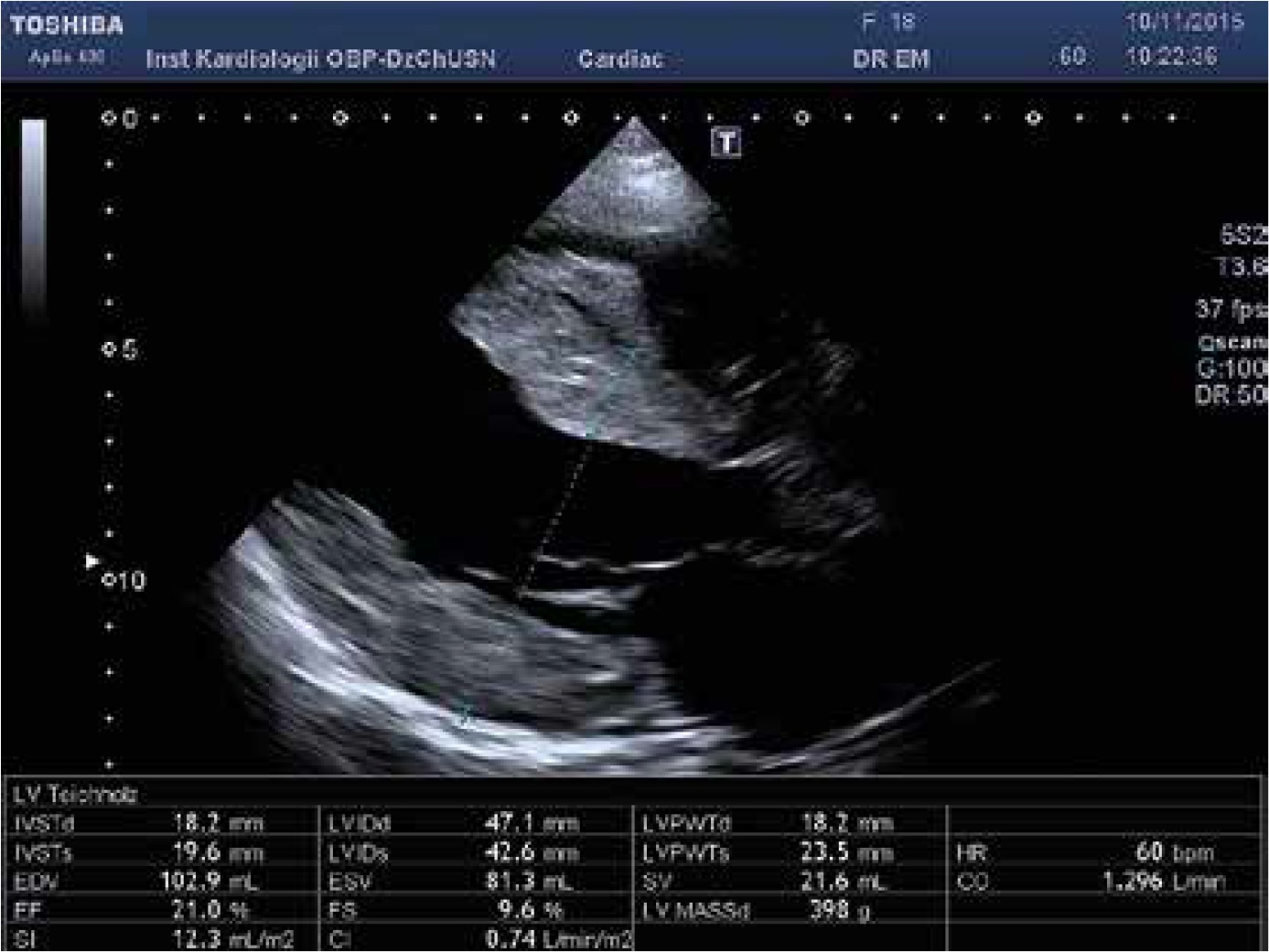
Figure 3
A – Apical 4-chamber view with speckle tracking. Low LV ejection fraction, systolic and diastolic asynchrony, decreased strain rate. B – Apical 4-chamber view and pulsed wave Doppler of transmitral flow – restrictive mitral flow pattern. C – Tissue Doppler imaging. Increased E/e′ ratio of 18.15 indicates high LV filling pressure. D – Modified apical 4-chamber view. Noncompaction of the right ventricular myocardium, the ratio NC layer/ C layer 14/4 mm
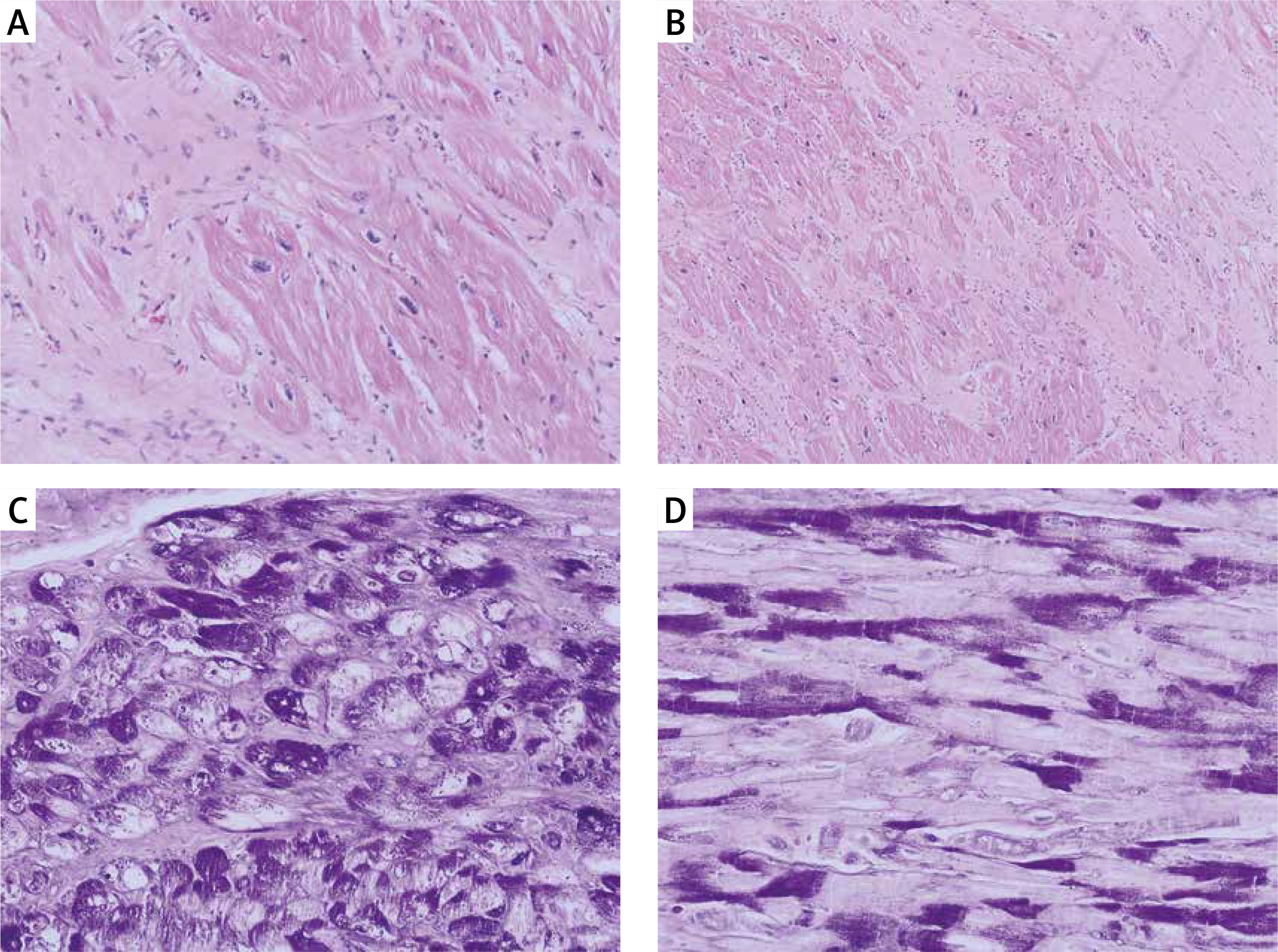
Figure 4
Microscopic picture of left ventricle showed cardiomyocyte hypertrophy with abundant eosinophilic cytoplasm and extensive interstitial fibrosis. Hematoxylin-eosin, original magnification: A – HE, 400×, B – HE, 200×. Additional histological stain revealed the presence of vacuoles containing glycogen, which is negative after diastase predigestion in some of the cardiomyocytes: C – periodic acid-Schiff (PAS) staining, original magnification, 400×, D – PAS with predigestion with diastase, original magnification, 400×
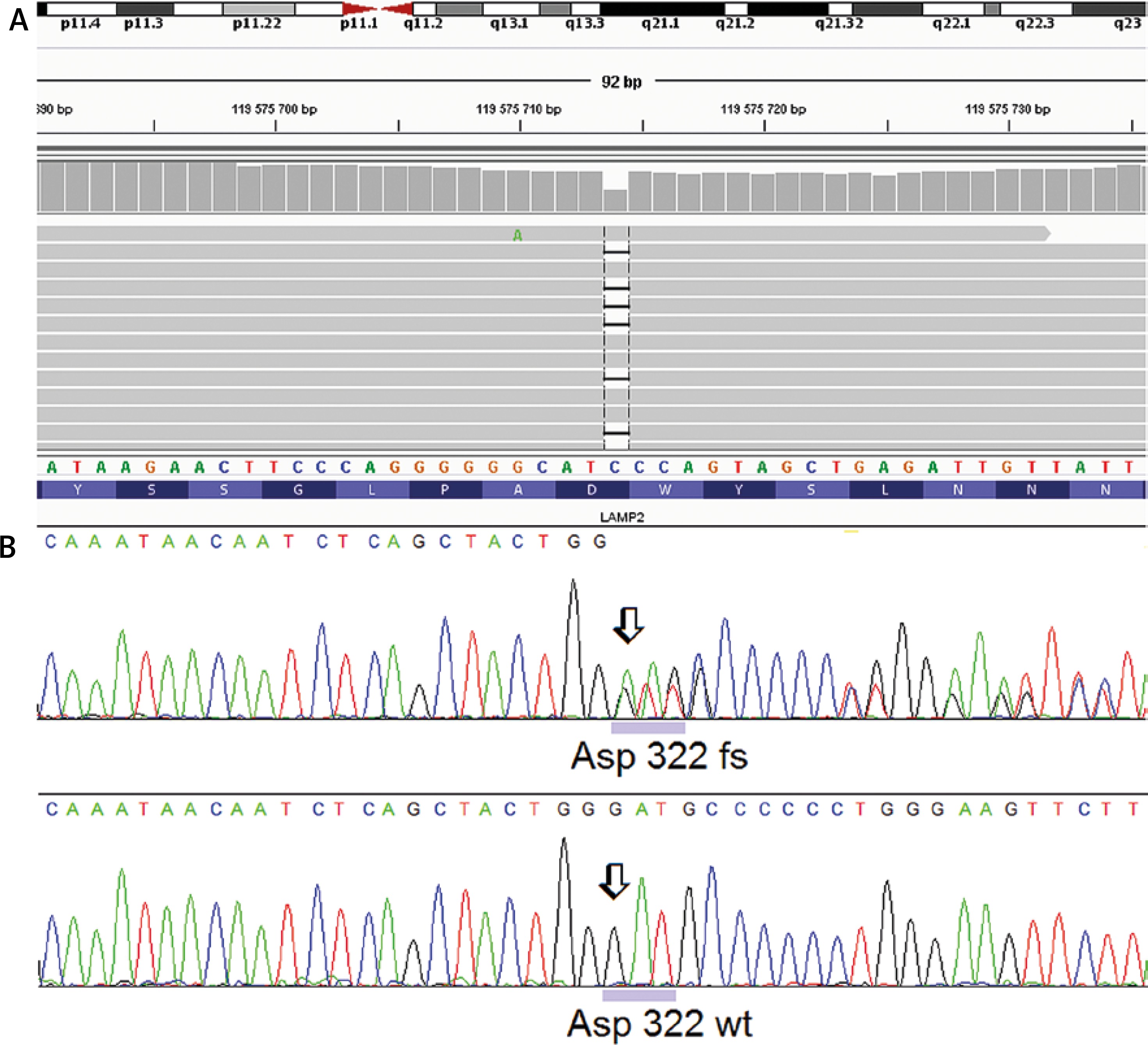
After receiving written informed consent the patient underwent genetic examination with the TruSight One Sequencing Panel (Illumina), which revealed a frameshift truncating variant in the LAMP2 gene, namely p.Asp322fs (c.964delG) annotated to transcript NM_002294.2 in exon 8 (Figure 5). Subsequent family screening with direct Sanger sequencing confirmed the presence of the truncating variant in the patient’s asymptomatic (based on normal non-invasive cardiac examination – ECG, echocardiogram, Holter 24 h ECG) mother. After careful analysis of the genetic test results of our patient we did not identify any other variants that could influence the clinical course of her disease.
Figure 5
Genetic analysis for lysosome-associated membrane protein 2 (LAMP2) gene. A novel frameshift mutation p.Asp322fs (c.964delG) annotated to transcript NM_002294.2 was identified in the proband and her asymptomatic mother. A – NGS result using IGV screenshot. B – LAMP2 frameshift deletion in the proband (Sanger sequencing) compared with wild type chromatogram below
The LAMP2 deficiency, due to mutated lysosomal membranous protein, results in accumulation of intracellular vacuoles containing glycogen and granular debris. In turn, this leads to myocardial hypertrophy, scarring and electrocardiological abnormalities [5]. Similarly as in our patient, the majority of mutations identified in Danon disease are predicted to cause LAMP2 deficiency [4]. Mean age at first presentation with Danon disease is lower in male patients than in female patients (11.7 vs 26.8 years respectively) [4]. Our proband developed the first symptoms at the age of 12 years. Presentation with hypertrophic cardiomyopathy dominates in men. A recent report revealed nearly equal rates of dilated and hypertrophic cardiomyopathy in women [6]. One third of male patients and 17.9% of women receive a cardiac transplant. There are also gender differences in age at cardiac transplant, namely 20.8 years in men and 40.2 years in women. Of interest, conduction abnormalities were reported in more than ¾ of men and women, but 68% of men and only 27% of women had a WPW pattern on electrocardiography. Our proband had a broad, fragmented QRS complex of 194 ms duration. Sudden unexpected death is of concern, and implantable defibrillators were placed in 41% of men and 31% of women [4]. An unexpected finding in our proband was the isolated noncompacted myocardium found in the right ventricle, as for now not associated with Danon disease. Isolated hypertrabeculation/noncompaction of the right ventricle is extremely rare and has been related to ventricular arrhythmia and sudden death [7]. Of interest, even left ventricular noncompaction was related to Danon disease once [8]. However, we cannot exclude that this finding is related to an unidentified gene. Magnetic resonance imaging is a useful tool in assessment of patients with hypertrophic cardiomyopathy and Danon disease, but we could not perform the examination because of early ICD implantation [9, 10]. Similarly to others, we have not found skeletal myopathy or mental retardation in our female carriers. In a recent report myopathy was found in 33% of women compared to 90% of men. Also, learning or cognitive disorders are reported in 100% of men, but only in 50% of women [4].
The presence of asymptomatic course of Danon disease among female obligate carriers of the LAMP2 mutations was observed earlier. Maron et al. published data on 7 women, obligate carriers of the LAMP2 mutations, who did not show abnormalities in clinical or noninvasive cardiological studies [11], similarly to the proband’s mother in our case. Gender differences in phenotypical expression may be explained by the fact that in male patients hemizygous for protein truncating LAMP2 mutations, there is a complete lack of LAMP2 protein, whereas in heterozygous females, expression of LAMP2 protein is expected in approximately 50% of the cells due to random X inactivation. One possible explanation is skewed X-inactivation in the proband, which could lead to increased mutation dosage, leading to a severe clinical picture compared to the mother, similarly as described in the case of female twins discordant for another lysosomal disorder, Fabry disease [12]. Also, mutation in regulatory elements influencing LAMP2 expression cannot be excluded: Smemo et al. recently found that the modularity in the cis-regulation of TBX5 can influence the phenotypic picture of Holt-Oram syndrome [13]. A particularly interesting hypothesis was postulated by Mathe et al. [14] and Oldfors et al. [15], suggesting that a detrimental inhomogeneous distribution of LAMP2 deficiency, rather than an overall reduction of LAMP2 protein, is of importance for the development of cardiomyopathy. We also cannot exclude the possibility that the variability of disease expression in our family is the result of mechanisms other than modifying genes.