Introduction
Coronaviruses are a large family of RNA viruses belonging to the Coronavirinae subfamily. Together with the Torovirinae they constitute the Coronaviridae family in the order of Nidovirales. The first member of the family of coronaviruses was described in 1932 in birds [1], and since the severe acute respiratory syndrome (SARS) outbreak in 2002–2003, new family members have been defined, highlighting the capabilities of coronaviruses to jump across species. They are enveloped RNA viruses with the longest RNA genome amongst all RNA viruses consisting of 26.2–31.7 Kb. They are constituted of four genera (α, β, γ, and δ– coronaviruses) out of which two (α and β) contain the seven subspecies that were known to be capable of infecting both animals and human hosts causing zoonosis. These are 229E-CoV, OC43-CoV, NL63-CoV, HKU1-CoV [2], the human enteric HECoV [3], the SARS coronavirus (SARS-CoV), and Middle East respiratory syndrome coronavirus (MERS-CoV). While most subspecies produce symptoms of the common cold and enteritis, SARS-CoV and MERS-CoV are capable of causing serious respiratory syndromes that in some cases can prove lethal [4].
Infection of the host cells by coronaviruses relies on the interaction of the viral particle with specific proteins on the cell surface. Interactions between the virus and host cells are initiated following the binding of the spike proteins with host cell receptors. Spikes are surface proteins of the envelope that give the crown-like appearance to the virus from which its name is derived. Coronaviruses and other budding viruses exploit the replicative machinery of host cells [5], shielded from host immune responses in double membrane vesicles [6, 7]. By budding through exocytosis for their egress, rather than inducing lysis or apoptosis and proinflammatory cytokine release, they aim to produce long-lasting infections with reduced associated pathogenesis. This is how they account for 15–30% of the annual common colds with mild symptoms, only occasionally affecting the lower respiratory tract [8, 9]. However, much of the pathogenesis of SARS-CoV-2 seems to be related to the cytokine storm resulting from the excessive activation of adaptive immune responses in vital organs and from dysregulation of the renin-angiotensin system (RAS) [10, 11]. The adaptation to infect the human host cells by exploiting the angiotensin-converting enzyme 2 (ACE2) receptor in SARS-CoV and SARS-CoV-2 was not without consequences: this has led to increased pathogenesis by disrupting RAS regulation through the antagonisation of ACE2. This is because ACE2 is a part of the depressor arm of the RAS, and it accounts for counteracting vasoconstriction, proliferation, oxidative stress, fibrosis, and pathogenesis of cardiovascular disease (CVD), all effects of the pressor arm [12]. Disruption of the depressor arm, the ACE2/Ang-(1-7)/MasR, and AT2R will lead to exacerbation of the effects of the pressor arm, the ACE/AngII/AT1R pathway, which seems to be responsible for the decompensation of pre-existent comorbidities in coronavirus disease 2019 (COVID-19) patients where the highest fatality is reported [13]. The resulting Ang-II increased levels lead to cell apoptosis with the release of proinflammatory cytokines [14]. This stimulates adaptive immune responses with the possibility of triggering a cytokine storm from its excessive activation [15].
Newly identified coronavirus SARS-CoV-2
On January 7, 2020, following a recent outbreak of a new type of highly contagious coronavirus [16], the eighth subspecies of human infecting coronaviruses was described and characterised while a test method was developed [17]. It was initially named 2019-nCoV [18] and later SARS-CoV-2. In a relatively short interval of 2 months, the outbreak turned into an epidemic that spread over several countries and continents infecting more than 100,000 individuals by March 7, 2020 [19]. On March 11, 2020 the outbreak was officially declared as a pandemic by the World Health Organisation (WHO) [20], infecting almost four and a half million people at the time of the writing of this manuscript [21].
The SARS-CoV-2, although distinct from its cousins MERS-CoV and SARS-CoV, is part of the same Betacoronavirus genre and Sarbecovirus subgenre with SARS-CoV but from a related subgenre with MERS-CoV, which is part of the Merbecovirus [22]. Although the disease caused by SARS-CoV-2 may be less severe than SARS and MERS, it seems more contagious, having a death toll already 180 times higher than the SARS and MERS epidemics combined [21].
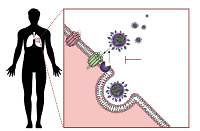
Coronaviruses are able to exploit many cell surface molecules – proteins and carbohydrates alike – in order to gain entry into target cells. Three receptors on the human cells have been shown to interact with the viral spike proteins that enable its fusion and incorporation within the host, and these include aminopeptidase-N, ACE2, and Neu 5.9 AC2. While aminopeptidase-N is the receptor used by the human coronavirus 229E-CoV [23], the sialic acid Neu5.9 AC2 is the preferred receptor for the human OC43-CoV. Angiotensin-converting enzyme 2 (ACE2), a type I integral membrane protein largely distributed in the vasculature, the endothelial cells of the heart and kidneys, brain, and lungs [24, 25], is the preferred receptor for NL36-CoV, SARS-CoV, and the new SARS-CoV-2, while MERS-CoV uses a different receptor – DDP4 (Figure 1) [26]. This distribution of the ACE2 in key organs might explain how patients with pre-existent cardiovascular system pathologies may be more susceptible to severe complications caused by the coronaviruses that use ACE2 to fuse with the human host cells, disrupting the physiological regulation of the RAS.
Implications of ACE2 in the RAS regulation
The receptor used by the SARS-CoV-2 to infect human cells, ACE2, is an important component in the regulation of RAS because it is part of the counteracting arm against the vasopressor pathway, ACE2/Ang1-7/MasR and AT2R. Accumulating evidence shows that the RAS is regulated differently in women and men, the androgens and oestrogens having different effects on the regulator arms of the RAS. Such differences allow the adaptation to increased total blood volume during pregnancy and confer a considerable degree of protection from cardiovascular disease in premenopausal women as compared to age-related men. This is done through the protecting arm of the RAS pathway, ACE2/Ang1-7/MasR and AT2R, which counteracts the vasopressor effects of the ACE/Ang II/AT1R axis. In men, the ACE/Ang II/At1R is predominant [27]. This might, at least in part, explain how lethality is lower in women than in age-related men and how exacerbation of pre-existent cardiovascular pathologies increase the lethality. Although these observations rely mostly on indirect evidence, a competitive mechanism may exist between RAS regulation through ACE2 and mechanisms of viral entry into the human host cells, because antagonism of ACE2 by the virus is associated with exacerbation of the effects of the pressor arm – the ACE/Ang II/AT1R axis.
In 2000 a new form of ACE was described, the ACE2 [28, 29], first characterised by Crackower et al. 2002 [30], involved in heart function and development of arterial hypertension. ACE and ACE2 have similar protein structures with small differences, which lead to different substrate specificities. ACE is the enzyme present in the lungs, which is responsible for converting angiotensin I to angiotensin II with vasopressor effects; ACE2 has been shown to exert counteracting effects on ACE action by inhibiting RAS through converting angiotensin I to angiotensin 1–9, which is further converted to angiotensin 1-7 by the ACE. ACE and ACE2 are thus functionally different enzymes with opposite roles [31]. SARS-CoV-2 spike glycoprotein binds to the cell membrane ACE2 to penetrate human cells. Further research confirmed the presence of ACE2 in the lung, heart, kidney, and vessel endothelium [32–34]. Zhao et al. showed that 83% of ACE2-expressing cells are alveolar epithelial; therefore, the lungs appear the most vulnerable target of SARS-COV-2 [35], while the heart, kidney, and central nervous system [36] are the second most affected organs by the viral burden. It has been demonstrated that the binding of the SARS-CoV-2 spike protein to ACE2 leads to ACE2 downregulation, which results in excessive production of angiotensin II by the related enzyme ACE, while ACE2 is not capable of converting angiotensin I to angiotensin 1–7. This results in increased pulmonary vascular permeability, and lung and heart injury [37]. The large distribution of ACE2 to other organs could explain the end-stage multi-organ dysfunction in severely infected patients [11, 38, 39], including neurological manifestations [36].
A China CDC weekly report through February 11, 2020 revealed the results of the demographic characteristics from 72,314 patient records admitted for the new coronavirus infection (COVID-19). The reported fatality in men was higher than in women (2.8% vs. 1.7%), high age and underlying cardiovascular pathologies being associated with increased fatality. Furthermore, synthetic datasets generated from this report showed that older women had a similar fatality to that of age-related men, making it possible for some sex hormone influences to affect the viral entry and pathogenesis, explaining some of the sex-/age-associated pathology differences in lethality and morbidity. The datasets created from this report suggest that patient’s advanced age was the most important risk factor of lethality from the disease, even greater than having any of the listed comorbidities [13]. These data rely mostly on indirect evidence because there is neither information about the correct prevalence of gender per age nor about comorbidities per age and gender in the CDC report [11]. At the current time, the reported sex ratio for the fatality men and women is still 1.6, but with higher fatality rates reported for men (4% in men and 2.5% in women) [40].
Differences in RAS regulation between men and women
Several lines of evidence show that intracellular RAS can operate independently of the circulating RAS, and there is a close interaction between sex hormones and RAS regulation. Oestrogens have been known to provide protective effects by modulating the RAS. One study on endothelial cells showed that the physiological levels of oestradiol increased ACE1 by 25%, but not ACE2 protein expression in vitro, through the oestrogen receptor alpha (ERα). The same study showed that while ACE2 protein expression remained unmodified following oestradiol exposure, the enzymatic activity of both ACE1 and ACE2 increased. Alternatively, the translation of ACE2 mRNA on protein could be down-regulated by oestradiol, directly due to increased ACE2 activity of increased Ang-(1-7) production. The increased activity of ACE2 without increased protein expression could be due to a direct oestradiol-induced ACE2 activity, but an indirect activation could not be confirmed [41].
Differences between men and women in RAS regulation could be attributed to a higher stimulation of ACE activity by androgens, as has been demonstrated in male mice [42], in which testosterone increases ACE activity, and also in women with hyperandrogenism from polycystic ovary syndrome [43]. This is further supported by sex differences in the regulation of arterial pressure and renal function by the RAS [44], with the balance tipped toward depressor pathways in women [45]. Moreover, women are protected against cardiovascular disease relative to men, prior to menopause [46]. Androgens are known to be involved in the sex differences in the regulation of arterial pressure, with oestrogen protecting against and testosterone exacerbating hypertension [44]. In the last two decades, special interest has been redirected towards the RAS with the discovery of additional receptors specific for angiotensin peptide fragments, suggesting the presence of a depressor arm of the RAS (ACE2/Ang(1-7)/MasR and AT2R), which counter-regulates the classical ACE/AngII/AT1R pathway [12].
Sex differences have been demonstrated in the regulation of blood pressure and renal function by the RAS. Significantly, the counter-regulatory arm of the RAS, including ACE2/Ang(1-7)/MasR and AT2R, is upregulated in females [27]. Oestrogen regulates all components of the RAS by increasing angiotensinogen synthesis while reducing the secretion of renin and synthesis of ACE [47]. In males, testosterone amplifies the pressor pathways of the RAS [48]. Regarding genetic sex differences, the SRY gene family, located on the Y chromosome in men, decreases promoter activity of ACE2 while upregulating promoter activity of ACE, angiotensinogen, and renin, respectively [49]. In addition, both the AT2R and ACE2 genes are located on the X chromosome, suggesting a greater role of these depressor RAS arm components in females. In males, the ACE/AngII/AT1R pathways are enhanced, whereas in females the balance is shifted towards the ACE2/Ang(1–7)/MasR and AT2R pathways. Evidence shows that premenopausal women are at lower risk of developing cardiovascular and renal disease as compared to aged-matched men, and this differential regulation of the RAS between men and women probably makes an important contribution. This cardiovascular protection in women lowers when reaching menopause and reaches the same incidence as seen in age-related men, probably being related to loss of oestrogen in the postmenopausal period. However, the mortality gender difference for COVID-19 is far more outspoken than the gender difference in the occurrence of CVD. This creates the premise of using endocrine therapy for the management of COVID-19.
Strategies of limiting viral entry into the human host cells and associated pathogenesis
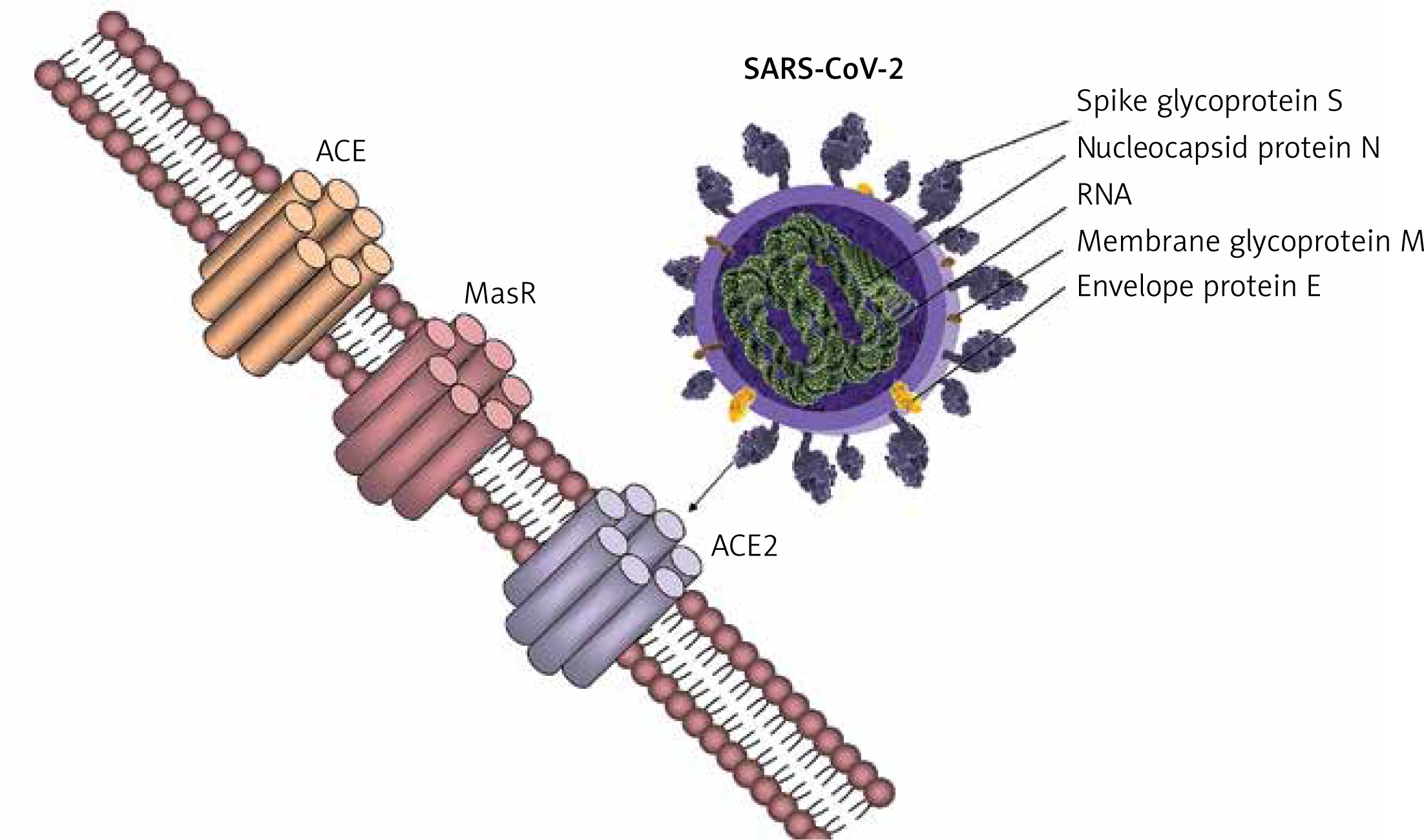
Several potential strategies of inhibiting viral entry into the human host cells to reduce lethality and associated morbidity are worth pursuing based on the ACE2 interaction with the SARS-CoV-2 spike proteins (Figure 2).
Figure 2
Competitive mechanisms involving viral entry into host cells and RAS regulation through ACE2. Physiological mechanisms of regulating the renin-angiotensin system involve the pressor pathway (ACE/Ang II/AT1R) and the counteracting arm – the depressor pathway (ACE2/Ang 1-7/MasR and AT2R). The new coronavirus (SARS-CoV-2) exploits ACE2 to ensure viral entry into host cells, antagonising the counteracting arm of the RAS system. This leads to complications associated with exacerbation of the ACE/Ang-II/AT1R axis, as has been observed in the pathogenesis of COVID-19. Possible strategies of limiting viral entry and pathogenesis involve modulating ACE2 expression and regulating the counteracting arm of the RAS system shifting the balance towards the ACE2/Ang 1-7/MasR and AT2R axis
Endocrine therapy in COVID-19
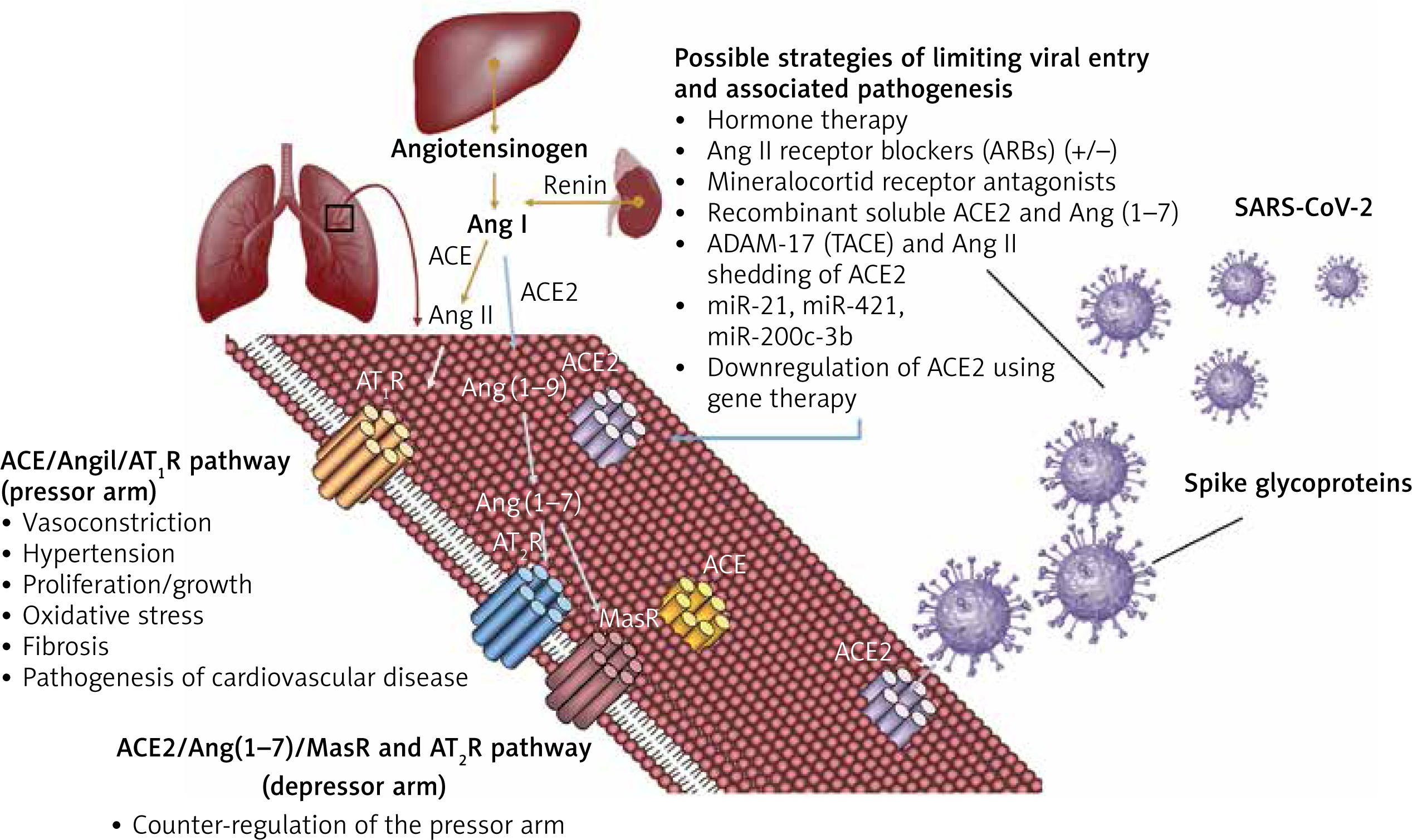
Regulation of the ACE2 could be attempted by several strategies, used alone or in combination. Endocrine therapy may prove useful in modulating ACE2 expression and regulating RAS in men and women. Androgen deprivation therapy (ADT) could potentially shift the RAS regulation towards the ACE2/Ang1-7/MasR and AT2R pathway in order to achieve a reduced lethality in men, at least to the extent reported in premenopausal women. To date, there is no study evaluating the effect of reproductive hormones and their effects on the infectious cycle of the SARS-CoV-2. This represents a direction worth pursuing because reproductive hormones (oestrogen 1 μg/ml and testosterone 3 μg/ml) were found to enhance the replication of the avian coronavirus in vitro, while progesterone had no effect. The same study found an enhancing effect of cortisone 3 μg/ml on the replication of avian coronavirus [50]. Another study on avian coronavirus showed that the adaptive immune response and cytokine activity was enhanced probably due to the effect of oestrogens in vitro [51]. In SARS-CoV and MERS-CoV, oestrogen receptor inhibitors were shown to be active against the infection in vitro [52]. However an in vivo mouse model showed that the severity of SARS is associated with enhanced accumulation of macrophages and neutrophils in the lungs leading to vascular leakage and alveolar oedema, and male mice had an increased susceptibility to these complications, independently of B and T cell response. Furthermore, ovariectomy or treating female mice with oestrogen receptor antagonists increased their lethality from SARS-CoV [53]. Together, these findings show that gender differences in the severity to SARS-CoV in murine models parallel those reported in patients and support the hypothesis of oestrogen receptor signalling as protective against COVID-19 in women. Although this may in part explain the reduced fatality from COVID-19 in premenopausal women, the implications of reproductive hormones in the pathogenesis of COVID-19 are even more compelling. Androgens have been shown to be a positive regulator of TMPRSS2 protease [54]. In lungs, TMPRSS2 (epitheliasin) is required for the activation of the spike viral protein for viral internalisation and activation of the fusion peptide (Figure 3), with alternative fusogenic mechanisms including furins, cathepsins, and other trypsin-like proteases, accounting for potential therapeutic targets in COVID-19 [55–58].
Figure 3
Schematic representation of the mechanism of viral entry into the lung cells by exploiting ACE2 and epitheliasin (TMPRSS2). SARS-CoV-2 enters the lung cells following the binding with the ACE2 and the activation of the fusion peptide in the spike protein by the membrane protease TMPRSS2. Androgen deprivation therapy decreases TMPRSS2 expression inhibiting the mechanisms of exploiting epitheliasin for the activation of the spike
The coding gene for the pulmonary epitheliasin is upregulated by androgen exposure, together with other genes involved in viral pathogenic mechanisms such as phospholipid metabolism, iron ion binding, oxygen binding, haeme binding, and clathrin-coated vesicle, and this was demonstrated in vitro on a culture of human lung adenocarcinoma-derived cell line – A549 [59]. The responsiveness of the TMPRSS2 gene to androgen stimulation was also reported in vivo in mice, and patient data correlated with these observations [60], while the implications of sex hormones in the immune response has been well documented [61]. Moreover, the protective effects of anti-androgens in SARS-CoV-2-infected patients was seen in an observational Italian study [62].
Based on these effects, androgen deprivation therapy (ADT), despite having the most well recognised but reversible side effects including hot flashes, loss of libido, and erectile dysfunction, is potentially beneficial for reducing lethality in men as well as women with hyperandrogenism from polycystic ovary syndrome [43]. While ADT represents an accessible option for aiding the treatment of COVID-19, a premise for its use should be the validation of these effects in controlled clinical trials including men, women, and even transgender patients on endocrine therapy.
Several ongoing clinical trials are currently investigating the interactions of corticosteroid hormone derivatives in COVID-19, such as methylprednisolone (NCT04263402; NCT04323592; NCT04244591), dexamethasone (NCT04263402; NCT04325061), budesonide, and formoterol (NCT04331470), while there are no clinical trials evaluating reproductive hormones in COVID-19. Testing endocrine therapy against SARS-CoV-2 could represent a close perspective. Nevertheless, several other strategies that can modulate the RAS and ACE2 expression are worth mentioning. The usage of recombinant ACE2 [63] could benefit COVID-19 patients because ACE2 might bind SARS-CoV-2 spike protein in the plasma before reaching other cells, rendering the spike incapable of binding other membrane ACE2, thus avoiding internalisation of the virus into cells. This is currently being evaluated in several clinical trials. (NCT04335136; NCT04287686)
RAS blockers in COVID-19
While using ACE inhibitor drugs that lower blood pressure may seem like a good approach to reduce the viral entry into the host, some of them are probably not useful in the treatment of COVID-19. Perindopril has little or no effect on ACE2 [64]. Captopril, enalapril, and lisinopril block ACE but not ACE2 and may be counterproductive. It is not clear if ACE inhibitors should be switched to other hypotensive drugs [65–67]. Knocking down the coding gene of ACE2 may seem like a good approach to blocking viral entry into the host cells, but this was shown in animal models to severely impair heart function [30]. However, angiotensin receptor blockers (ARB) as well as mineralocorticoid receptor antagonists (e.g. spironolactone) increase both ACE2 levels and Ang (1–7) levels [68, 69]. There are sufficient data showing that ARB treatment results in ACEs upregulation in humans and rats: losartan and olmesartan were demonstrated to increase cardiac ACE2 expression three-fold following 1 month of treatment after myocardial infarction in rats [70]. Losartan was also demonstrated to upregulate renal ACE2 expression in mice [71]. Olmesartan used in patients with hypertension induced high urinary ACE2 levels [72]. Therefore, higher ACE2 expression after treatment with ARBs, while seeming paradoxical, might protect against lung and heart injury. This may be the result of blocking the excessive angiotensin caused by the SARS-CoV infection, as well as, most importantly, upregulating ACE2, with increased production of angiotensin 1–7 [73]. A recent article suggested that ARBs might be beneficial for patients infected with SARS-CoV-2 [74]. Several ongoing clinical trials are currently investigating the impact of RAS inhibitors in COVID-19 (NCT04335123; NCT04337190; NCT04331574; NCT04330300; NCT04312009; NCT04311177; NCT04335786) and are looking for arguments to continue or discontinue RAS inhibitors (NCT04338009). A possible method of treatment that would probably be resistant to SARS-CoV-2 mutations is to use available ARBs, such as losartan, telmisartan, and olmesartan to reduce the binding of the virus to ACE2 and decrease the aggressiveness and mortality from virus infections [75]. However, drugs that interfere with the regulation of the counteracting arm of the RAS directly or indirectly have been shown to induce an increase in ACE2 expression, and this was observed for ARBs [70, 76], statins [77–80], and the propionate derivative ibuprofen [81] in preclinical studies. Whether this increase is detrimental due to increasing binding sites for the virus or actually beneficial, being part of the mechanisms of reducing the deleterious effects of the pressor arm of RAS, remains elusive and requires further investigation. A recently published analysis found no association between ARBs or ACEI use and COVID-19 test positivity in a cohort of 18,472 patients tested for COVID-19 in a single healthcare system [82]. In the absence of clinical evidence to the contrary, the clinical consensus is to advise patients not to discontinue ACE inhibitors or ARBs in the setting of the COVID-19 pandemic [66, 83].
Other strategies of inhibiting viral entry in COVID-19
Other strategies of reducing ACE2 include ADAM metallopeptidase domain 17 (ADAM17), also known as tumour necrosis factor-α-converting enzyme (TACE), capable of shedding ACE2 from the cells through AngII [84] and miRNAs that are in turn capable of regulating ACE2 expression in the lungs (miR-200c-3p [85], miR-21 [86], miR-421 [87]), while restoration of these RAS depressor pathways in older women or upregulation of these in males and females by gene therapy using adenoviruses for blocking angiotensin receptors could represent a therapeutic intervention to assist the treatment of SARS-CoV-2 infected patients. Using such strategies could be followed by a destabilisation of the blood pressure and should be done under specialised cardiological observation.
An efficient active immunotherapy (vaccination) would be the cornerstone against COVID-19, but it could take many months to develop. This may also be limited in its efficacy or may even prove ineffective because viral antigens can regularly change with adaptation mechanisms. Moreover, pre-existent antibodies from a recovered infection or vaccination may progressively fade away [88, 89] or may not exert sufficient protection against re-exposure to human infecting coronaviruses, as shown in studies of SARS-CoV and MERS-CoV [90, 91]. Passive immunotherapy with monoclonal antibodies against antigens of the spike protein could represent a closer perspective, and this was previously achieved in SARS-CoV in vitro [92].
Other treatment options in COVID-19 are under evaluation, and while preliminary results seemed promising with antimalarials [93] and replicase inhibitors [94], several recent studies failed to show clinical benefits [95, 96], emphasising the need to evaluate other potential therapeutic strategies against SARS-CoV-2.
Conclusions
While we may assert that there are multiple potential strategies against the early steps of viral entry into the human host cells and the pathogenesis associated with the dysregulation of the RAS, evaluation of the effects of such agents in large cohorts is required to validate their efficacy in COVID-19. Drug repurposing can prove beneficial in controlling the extent of the disease while a specific treatment is being developed, and exploiting endocrine therapy against SARS-CoV-2 could represent a unique approach, which is worth pursuing given the rapid spread of the pandemic. However, drug combinations may prove more effective than single agents, and combining endocrine therapy with other strategies of inhibiting viral entry could represent a close perspective in the current pandemic because active immunisation might require many months before being accomplished and readily available worldwide.