Introduction
Hypertriglyceridemia (HTG) is the most common lipid disorder, affecting approximately 10% of the world’s population [1, 2]. The diagnosis of HTG depends on the fasting triglyceride levels. Based on the Endocrine Society Clinical Practice Guideline, HTG is classified as mild (triglycerides (TG) 1.7–2.2 mmol/l; 150–199 mg/dl), moderate (TG 2.3–11.2 mmol/l; 200–999 mg/dl), severe (TG 11.3–22.5 mmol/l; 1000–1999 mg/dl), and very severe (TG ≥ 22.6 mmol/l; > 2000 mg/dl) [1, 2]. Familiar non-syndromic HTG is caused nearly exclusively by biallelic defects of the following five major genes: LPL, APOC2, APOA5, LMF1, and GPIHBP1. Familial LPL deficiency is the most common form of familial hypertriglyceridemia and accounts for nearly 95% of all cases [3–7].
Biallelic pathogenic variants in the LPL gene are associated with familial lipoprotein lipase (LPL) deficiency (MIM #238600) [8], a disease manifesting with recurrent pancreatitis, abdominal pain, cutaneous xanthomata, and hepatosplenomegaly. At the molecular level, biallelic pathogenic variants in the LPL gene impair clearance from the chylomicrons to the plasma. The inheritance pattern of the disease is in general autosomal recessive. Heterozygotes are mostly asymptomatic but may have mild to moderate hypertriglyceridemia and may develop atherosclerosis at a younger age. The inheritance pattern of the familial lipoprotein lipase deficiency is also sometimes described as a codominant inheritance mode, where carriers of one allele of the recessively inherited gene exhibit only mild, often nonspecific symptoms and usually a late disease onset, whereas those homozygous for the pathogenic LPL variant present with a full disease phenotype [9]. In contrast, some diseases show both recessive and dominant inheritance, such as myotonia congenita (Thomsen myotonia with CLCN1 pathogenic variants, dominant inheritance; MIM #160800 and Becker myotonia with CLCN1 pathogenic variants, recessive inheritance, MIM #255700) [10] or calpainopathy (traditional recessive inheritance MIM #253600 and recently described dominant inheritance; MIM #618129) [11].
Since the beginning, the carrier status of the pathogenic/likely pathogenic LPL variants has been widely discussed. In one of the first studies investigating large pedigrees with heterozygous carriers, the LPL level, although decreased by about 50%, did not make it possible to differentiate between carriers and non-carriers with similar phenotypes and the same lipid levels occurring in both groups [12]. Carriers presented a pathogenic plasma lipid profile and lipid changes including increased TG, very-low-density lipoprotein (VLDL), and apolipoprotein B (apoB), and decreased low-density lipoprotein (LDL) and high-density lipoprotein (HDL). Interestingly, these differences have been mainly observed only in probands older than 40 years, suggesting a contributory role of secondary and environmental factors, such as obesity and insulin resistance. Available studies have not been conclusive regarding the role of the pathogenic variants associated with monoallelic hypertriglyceridemia and the intermediate phenotype [13, 14].
Some studies have reported that heterozygous carriers may present a range of HTG levels rather than a binary affected/non-affected status. In a cohort of 15 LPL pathogenic heterozygotes, the HTG level varied greatly, and 14.8%, 67.0%, and 18.2% were in the normal, mild-to-moderate, and severe HTG ranges, respectively, in the 1.5-year longitudinal studies [13]. In our group of Polish patients, we confirm not only the range of phenotypes but also a near-to-biallelic phenotype in heterozygotes and their parents, suggesting a dominant inheritance pattern.
Material and methods
Whole exome sequencing (WES) was performed on 5623 Polish individuals (as of 23.05.2024) In 52 cases the indication for WES genetic testing was “hypertriglyceridemia” and for 5571 there was another clinical indication, mainly autism spectrum disorder, dysmorphia and neurodegenerative diseases. Technical sequencing was performed by CeGaT (Tübingen, Germany). Bioinformatic analysis and scientific and diagnostic interpretation of variants were performed by MEDGEN laboratory (Warsaw, Poland). The genomic DNA of patients was extracted from a whole blood sample using Prepito. The sequencing exome library was prepared according to Twist Exome Enrichment (Twist Bioscience). The enriched DNA libraries were sequenced by the Illumina NovaSeq 6000 instrument, 2 x 100 bp.
Raw sequencing reads were mapped to the human reference genome GRCh37 and GrCh38 assembly using BWA MEM (bwa-mem2.avx2 mem 0.7.17-r1188) [15]. Duplicates were removed using biobambam2 version 2.0.183 [16]. Variants were called using HaplotypeCaller (GATK v4.2.6.1 [17, 18]) and FreeBayes v1.3.2 [19], and named using Variant Effect Predictor (VEP109) [20]. The variants were classified according to ACMG recommendations [21]. The presence of the variant in control populations was checked in 1000 Genomes [22] and gnomAD (v4) (Broad Institute) databases [23]. The in silico splicing analysis was performed using algorithms embedded in Alamut Visual Plus software (Sophia Genetics), i.e. SpliceSiteFinder-like, MaxEntScam, NNSPLICE and GeneSplicer and SpliceAI [24]. The presence of the detected pathogenic/likely variants was confirmed by Sanger sequencing. The XHMMv1.0 algorithm and in-house scripts were used to search for small, rare, hemizygous and homozygous copy number variant (CNVs).
All exomes were searched first for variants in the LPL gene reported in the ClinVar database as “pathogenic/potentially pathogenic” regardless of reported clinical manifestations. Then, our entire WES database was searched for rare nonsense variants, frameshifts and canonical splicing, and large deletions and duplications not reported in the ClinVar database. For patients included in our study, pathogenic rare variants were only found in LPL and none of the other canonical familial chylomicronemia syndrome (FCS) genes (i.e. APOC2, APOA5, GPIHBP1 and LMF1) [25].
Results
Genetic findings in the Polish cohort
The tested individuals were 1–76 years old (Table I). In the WES analysis of individuals with = “hypertriglyceridemia’’ as an indication for testing, there was a pathogenic/likely pathogenic LPL variant present in 23% (12 out of 52). In the WES analysis of individuals with “other (not hypertriglyceridemia)” as an indication for testing, there was a pathogenic/likely pathogenic variant in 0.18% (10/5571) (p < 0.05).
Table I
Variants, zygosity, TG levels, and approximate age at molecular diagnosis in patients with pathogenic LPL variant
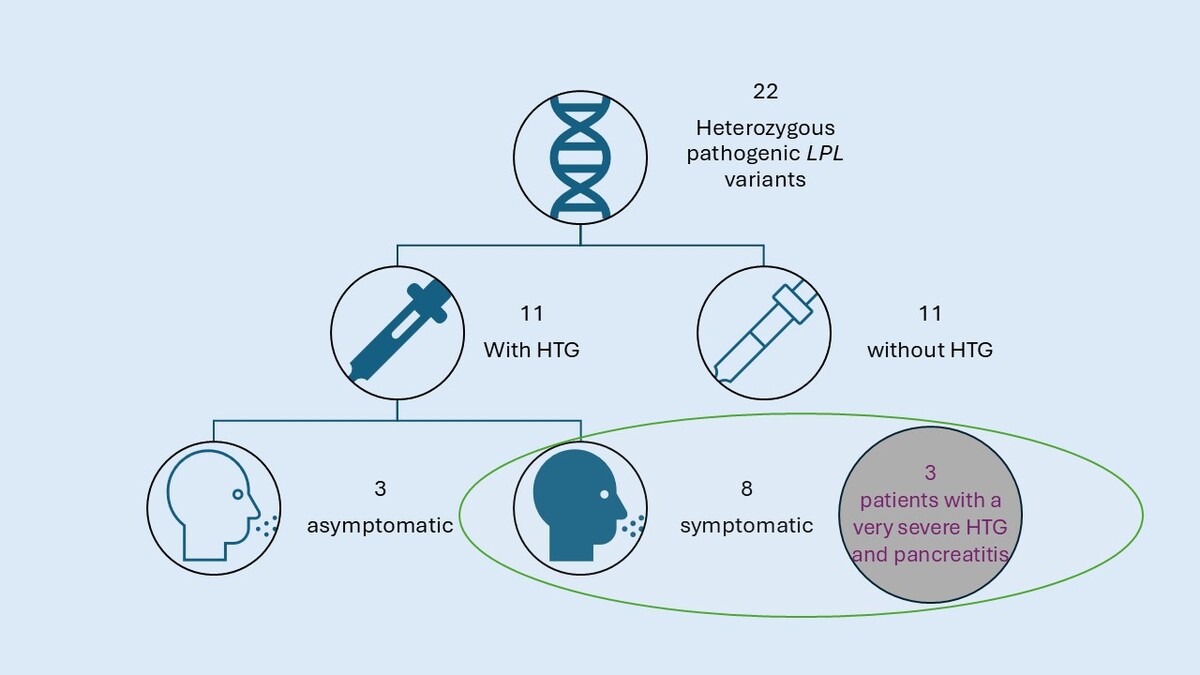
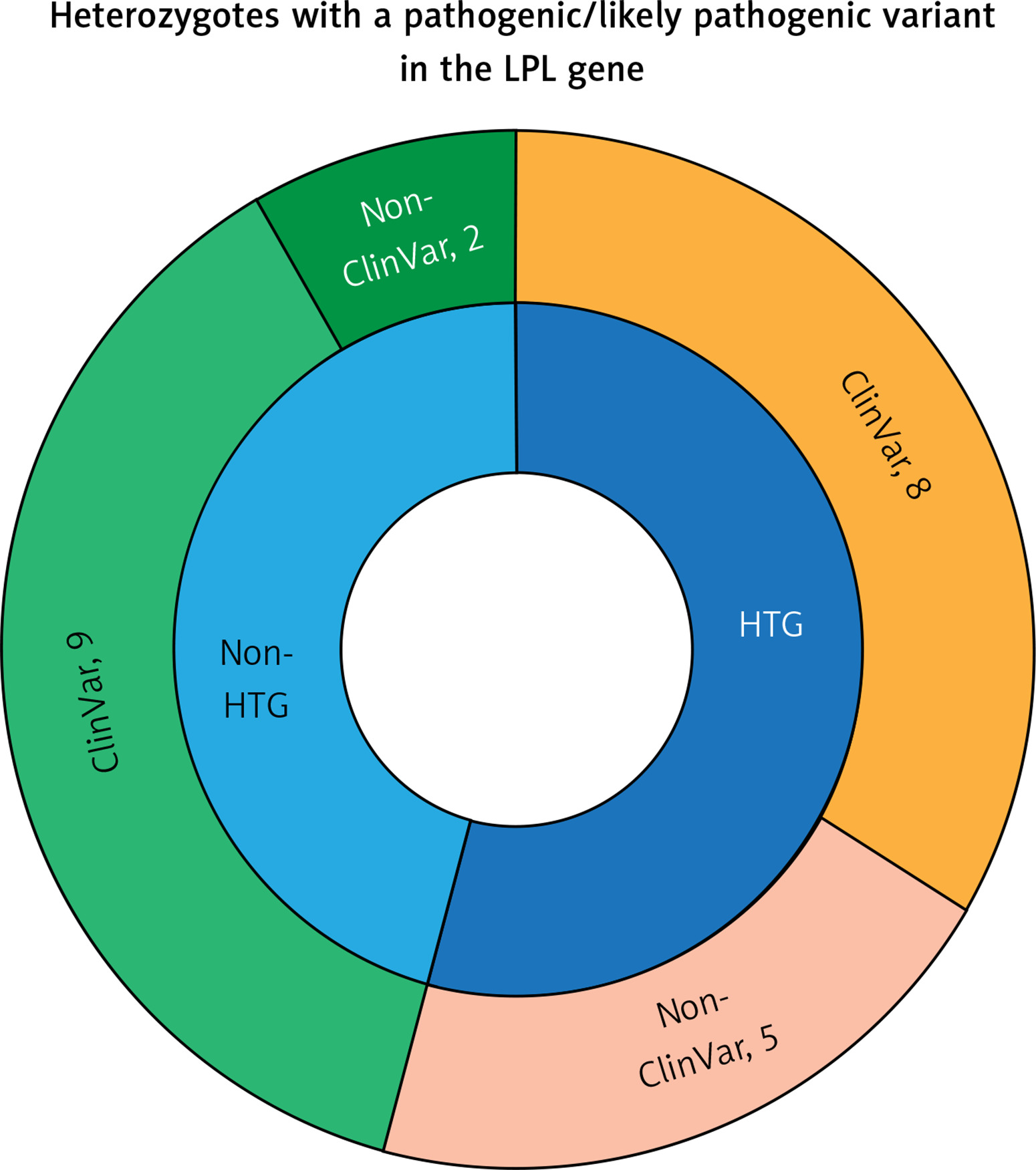
In 17 individuals the variant(s) in the LPL gene have been reported as pathogenic in ClinVar and in 7 cases the variants have not been reported as pathogenic in ClinVar, but fulfilled the pathogenicity criteria according to the ACMG classification (Table I, Figure 1). One patient was homozygous and one compound heterozygous for the pathogenic LPL variants. Interestingly, in 2 cases there was a CNV in the LPL gene causing duplication of the whole exome 6 and predicted to be pathogenic according to ACMG criteria. Thirteen individuals were diagnosed with HTG, whereas 11 were not, despite carrying a pathogenic variant.
Figure 1
Distribution of the pathogenic/likely pathogenic (according to the ACMG criteria) Clin- Var and non-ClinVar variants in the heterozygotes with and without HTG
In the group of patients with a known pathogenic variant, 15 were heterozygous, 1 was homozygous, and 1 was a compound heterozygous.
Phenotype analysis
In 11 cases the heterozygotes for the pathogenic LPL variant did not present elevated TG levels, whereas in 11 cases they did. We then investigated common LPL variants: LPL p.Trp113Arg and LPL p.Gly215Glu. Two LPL p.Trp113Arg carriers had HTG (age 11 and 76) and 3 normal HTG levels (age 9, 9, and 1 year). One LPL p.Gly215Glu carrier showed HTG (age 1 year) and three normal TG levels (age 6, 11, and 18 years) (Table II).
Table II
TG levels divided by variant
| Variant | TG normal | HTG |
|---|---|---|
| For all pathogenic variants | 11 | 11 |
| p.Gly215Glu | 3 | 1 |
| p.Trp113Arg | 3 | 2 |
Interestingly, not only HTG has been found in heterozygotes, but also 8 monoallelic patients had clinical symptoms, including non-specific symptoms such as abdominal pain and periodic nausea (Table III). Three out of 8 patients had at least 1 episode of acute pancreatitis in the history, while 3 had enlargement of the liver and/or spleen. None of these symptomatic patients drank alcohol. One patient had only moderately high triglyceride levels without additional symptoms typical of LPL deficits. There was a correlation between the presence of the HTG and age in the group of heterozygous LPL individuals. The medium age in the heterozygotes with HTG was 24.5 years (2–76 years), whereas in the heterozygotes without HTG it was 12.8 years (1–59 years).
Table III
Phenotypic features of individuals with HTG and heterozygous pathogenic LPL variants
We identified 3 individuals with a heterozygous LPL pathogenic variant (according to the ACMG criteria) and very severe HTG (TG ≥ 22.6 mmol/l; >2000 mg/dl) (Table I). In 2 cases the TG level exceeded 3000 mg/dl. All of the individuals presented with pancreatitis – a symptom related to HTG. One of the patients even suffered from 3 episodes of severe pancreatitis. One individual with severe/very severe HTG had a normal BMI, one was mildly overweight (BMI 25.3 kg/m2) and one was obese (BMI 36.3 kg/m2) (Table III). All these three individuals were older than 40 at the time of the genetic testing. Two variants were loss-of-function (LoF): one was a duplication of the whole exome 6, and one was classified as a missense. All variants were located in exons 5 and 6 of the LPL gene. None of the variants has been reported in ClinVar, and all variants were classified as pathogenic according to the ACMG classification.
Segregation analysis was performed in all three cases. In 2 cases, nobody from the family inherited the variants. In patient 3, a daughter (born in 2009) affected with the same deletion showed TG of more than 700 mg/dl in the 2nd decade of life already.
Discussion
We described the phenotypic and molecular spectrum of a large cohort of homo- and heterozygous individuals for pathogenic variants in the LPL gene. We found that individuals with a heterozygous LPL variant present the whole range of phenotypes, from being asymptomatic to displaying the severe, fully developed phenotype with very severe HTG and clinical symptoms. We cannot exclude that the development of the phenotype is influenced by factors such as sex, age, environment, and comorbidities.
The heterozygotes with HTG were older than those without HTG; however, there was no age threshold for symptoms of heterozygous individuals with very severe HTG presenting already at the age of 2. For example, the variant LPL p.Gly215Glu was reported in a young child (2 years old) with severe HTG, whereas older children with the same variant were asymptomatic (Table I) – raising a question about the influence of environmental exposure. This is in contrast to the study by Perera et al. [13], where only one individual out of 15 had HTG below the age of 40. There was no correlation between the type of variant (LoF or missense) and disease severity/presence of HTG in the laboratory testing. This is in opposition to some studies that suggested a more severe phenotype in the LoF variants than in missense variants in the LPL gene [13, 26, 27]. Two patients with very severe HTG presented with LoF variants. In 1 case, we report a duplication of exon 6. Heterozygous deletions in the LPL gene have been already associated with severe, but not very severe HTG [28, 29]. In a few patients in the literature, duplications disrupting exon 6 have been associated with very severe HTG, but without any symptoms related to it, such as pancreatitis; however, no genotype-phenotype correlation could be established [29].
We confirm that the heterozygous LPL carriers may present with the full range of phenotypes. This is in accordance with the recent approach not to classify the disease as dominant and recessive but rather as a heterozygosity spectrum. Additionally, we found that the LPL heterozygotes may exhibit very severe HTG in laboratory testing and show clinical manifestations and dominant variant inheritance patterns. The inheritance pattern of the LPL gene is described either as recessive or codominant, meaning that two pathogenic variants determine a more severe phenotype while heterozygotes for the pathogenic variants in the LPL gene have an intermediate phenotype. We argue that LPL could also follow a dominant inheritance pattern with affected parents passing the variant related to the phenotype to their offspring. It should not be forgotten that the development of the phenotype is certainly influenced by factors such as age, sex, and comorbidities (Table III).
For the first time, we report the frequency of the heterozygous pathogenic/likely pathogenic LPL variants in the Polish population, estimated at 1 : 1000 individuals. An important fact is that the majority of samples for the WES examinations derive from the MEDGEN laboratory with the indication for intellectual disability, autism, dysmorphia, and neurodegenerative diseases. Because mainly these indications are the focus of referring physicians, they often forget to enter a symptom such as ‘hypertriglyceridemia’, considering it less clinically relevant. It is therefore possible that there are indeed more of these symptomatic LPL heterozygotes, as this group is not fully characterized clinically. It is important to remember that because of the indications for testing, including HTG and hypercholesterolemia, that there is an overrepresentation of HTG in our cohort, so it is not representative for the whole Polish population. In the future, it would be advisable to study a random population group.
Our study has several limitations. Some of them are related to the technical limitations of the methodology. Whole exome sequencing can detect only coding sequences, which leaves a possibility that there is a structural or a splice site variant that has been omitted, especially in severely affected heterozygous individuals. Another technical limitation is that the deletion of exon 6 has not been confirmed by the second molecular method. Secondly, we have only limited phenotypic data. We do not have exact lipid levels for individuals without pathogenic LPL variants identified, which may be a source of bias. Also, we do not have full data on the environmental and secondary factors, influencing LPL levels. Especially information on the BMI is missing in several cases, which may be a source of bias. We do not have a longitudinal follow-up, so we cannot reliably describe the age of onset of the HTG and age when HTG develops, which means we cannot rule out that the asymptomatic heterozygotes will become symptomatic at a later point of life. Additionally, we did not investigate a polygenic risk score in the heterozygotes. Polygenic accumulation of genetic risk is associated with a mild [30], but also severe HTG disease form [31]. Common variants account for most of the mild HTG cases [32], but also for around 46% of cases of the severe disease form [30].
In conclusion, we argue that although the individuals carrying the single LPL pathogenic/likely pathogenic variant display the whole disease spectrum, the severe phenotype of heterozygotes with dominantly inherited LPL-related HTG may also exist. There was no clear association between the type of variant and the onset of the disease. Phenotypic and demographic characterization of further heterozygotes should help to better understand the background of the symptomatic spectrum.