Introduction
Cardiovascular diseases are the leading cause of death in developed societies. Extensive preventive measures in many countries have managed to reduce the incidence of coronary heart disease and myocardial infarction. Heart failure still remains the real challenge, and can be called the epidemic of the 21st century [1]. The major processes involved in the progression of myocardial dysfunction are adrenergic stimulation and inflammation. Chronic stimulation of the β-adrenergic receptors leads to augmented preload and afterload, increases heart rate and promotes apoptosis [2]. Clarification of the changes that agonists of β-adrenergic receptors and proinflammatory cytokines exert on the heart muscle can contribute to improving the care of patients with cardiovascular disease.
Interleukin-6 (IL-6) is one of the first described pleiotropic cytokines involved in a number of physiological processes [3]. Despite numerous experimental and clinical studies, the effect of IL-6 on the development of cardiac insufficiency has not yet been fully understood and explained. It is believed that a high concentration of IL-6 can contribute to the development of heart failure [4] and is an indicator of worse prognosis in patients with cardiovascular disease [5]. Expression of IL-6 increases in the mouse and rat myocardium after ischemia and reperfusion [6, 7], in the course of cardiac hypertrophy or pressure overload [8, 9]. Excessive IL-6-dependent signaling in the mouse model of myocardial infarction promotes cardiac inflammation, adverse remodeling, and heart failure [10]. Stimulation of rat cardiomyocytes with IL-6 reduces their contractility through enhanced synthesis and activity of inducible nitric oxide [11]. In patients with acute myocardial infarction IL-6 is a marker of more severe myocardial damage and ischemia and may reduce myocardial contractility [12]. On the other hand. many reports suggest involvement of IL-6 in compensatory hypertrophy of myocardium, neovascularization and cardioprotection [13], and IL-6 seem to be crucial for ischemic preconditioning and for attenuation of acute inflammatory responses within the myocardium during septic shock [14–16].
The ambiguous role of IL-6 in cardiovascular disease is probably the result of complexity of its downstream signaling pathways. IL-6 exerts its biological activity via its receptor, which is composed of two molecules: IL-6 specific subunit gp80 (IL-6 receptor) and intramembrane gp130, a signal transducing subunit common for all cytokines from the IL-6 family. Binding of IL-6 with membrane-bound gp80 leads to homodimerization of gp130 and activation of JAK kinases, which phosphorylate tyrosine residues on the cytoplasmic domains of the gp130 molecules at Y765, Y812, Y904 and Y914, leading to subsequent phosphorylation of STAT3 (signal transducer and activator of transcription 3). Activated STAT3 molecules form homodimers or heterodimers with STAT1, and rapidly translocate to the nucleus [17, 18].
There is however also a possibility to transduce the signal from other gp130 signaling cascades. Phosphorylation of Y757 of the gp130 protein allows for binding the cytoplasmic tyrosine phosphatase Shp2 (protein tyrosine phosphatase – PTP) [19], which further activates Ras protein and subsequently stimulates MAPK (mitogen-activated protein kinases) and PI3K (phosphatidylinositol 3-kinase) cascades [18, 20]. Phosphorylation of gp130 at the same tyrosine residue forms a docking site also for SOCS3 (suppressor of cytokine signaling 3) protein, which competes with Shp2 for the binding site, and attenuates gp130-dependent activation of MAPK and PI3K pathways [21]. Although JAK/STAT is considered the major transduction route activated by IL-6, also MAPK and IP3K dependent signaling greatly contributes to its biologic effects [21].
The cellular effects of IL-6 are modulated by intracellular molecules of SOCS and PIAS (protein inhibitors of activated STAT) families. Both these groups of proteins are negative regulators of IL-6-dependent pathways. As stated above, SOCS3 can bind the gp130 intracellular domain, and thus inhibit binding of JAK protein kinase activity, or by inhibiting JAK kinases directly [22]. The expression of the SOCS3 gene is induced by IL-6, IL-10 and interferon-γ [23]. PIAS1 and PIAS3 exert an inhibitory effect by direct binding to STAT1 and STAT3 and inhibiting their nuclear translocation [24].
Both IL-6 and increased sympathetic activity are upregulated in diminished cardiac function and share a few common intracellular pathways (MAPKs) [25]. Excessive stimulation of β-adrenergic receptors upregulates expression of IL-6 in the cardiomyocytes and cardiac fibroblasts [26–28], so at least some overlapping activities of both these factors could be expected. Additionally, in our previous study short term β-adrenergic stimulation with isoproterenol (ISO) was able to induce STAT3 phosphorylation independently from IL-6 [29], supporting the hypothesis assuming the interplay between IL-6- and β-adrenergic-dependent signaling pathways. However, this relationship is not conclusively determined and its better understanding may reveal new mechanisms involved in myocardial remodeling and secondary cardiac injury in the course of chronic heart failure.
The aim of the present study was to clarify the role of IL-6 in activation of selected intracellular signaling pathways induced by chronic β-adrenergic stimulation with isoproterenol.
Material and methods
Ethical approval
The experimental procedures were carried out according to the European Council Directive of 24 November 1986 (6/609/EEC) and were approved by the Local Ethics Committee, decision no. 59/2008.
Animals
Experiments were performed on 16 male mice C57BL/6JIL6‑/‑TMKopf (IL-6 KO) (body weight 27.9 ±2.4 g) and 17 wild male mice C57BL6J reference strain (WT) (body weight 27 ±2 g), aged 12 weeks. Animals were housed in an air-conditioned room at a temperature of 22 ±1°C and humidity of 65% in a 12-hour day/night cycle starting at 07.00.
Eleven WT and ten IL-6 KO animals of both genotypes received intraperitoneally a solution of ISO (Sigma) (50 mg/kg body weight dissolved in 0.9% NaCl) once a day for 16 days. Remaining animals of both genotypes received an intraperitoneal injection of 0.9% saline solution for 16 days. Animals were sacrificed by cervical dislocation 24 h after the last injection in order to avoid any direct effects of administered ISO on the analyzed transduction pathways. Hearts were dissected, apical parts of left ventricles were cut into two sections, frozen in liquid nitrogen and then placed at –80°C, whereas heart bases were fixed in phosphate buffered formalin. The genotypes were assessed using standard PCR of DNA isolated from mouse tails.
Western blotting
Frozen tissue samples were homogenized on ice in radio-immunoprecipitation assay buffer (Sigma, R 0278) containing 1 mM dithiothreitol, 0.5 mM sodium fluoride, 0.2 mM sodium orthovanadate, 0.5 M EDTA with addition of protease inhibitor cocktail 1 : 100 (Sigma, P8340), then centrifuged. Protein concentration in the supernatant was measured using the Bradford method [30]. Protein separation was performed under reducing conditions using sodium dodecyl sulfate polyacrylamide gel electrophores on 12% acrylamide gel (Bio-Rad, 161-0154). Then proteins were blotted onto 0.45 μm pore-size nitrocellulose membrane (Bio-Rad) in buffer containing 48 mM Tris, 39 mM glycine, 20% methanol, 0.037% sodium dodecyl sulfate) under constant current 320 mA for 90 min. Then the membrane was rinsed with deionized water and incubated in Miser Antibody Extender Solution NC (Thermo, 32110). Then blots were incubated in blocking solution containing 5% bovine serum albumin/Tris-buffered saline with 0.1% Tween 20 – 0.1% TBST) and probed with the following primary antibodies pERK1/2 (Thr202/Tyr204, Cell Signaling (cs)#9101), ERK1/2 (cs#9102), pSTAT3 (Tyr 705, Santa Cruz (sc) -71792), STAT3 (sc-7179) p-c-Raf (Ser289/296/301, cs#9431), c-Raf (cs#9422), p-p38 (Thr180/Tyr182, cs#9211), and p38 (cs#9212), which were also prepared in the same blocking solution. For incubation with primary antibodies – SOCS3 (sc-9023), PIAS1/3 (Protein inhibitor of activated STAT, sc-271172) and α-tubulin (sc-5286) –membranes were blocked and primary antibodies were dissolved in 5% non-fat milk in 0.1% TBST. Then blots were rinsed with 0.1% TBST and incubated with secondary antibodies conjugated to horseradish peroxidase (Serotec-STAR54 and Sigma-A9303). Blots were visualized using enhanced chemiluminescence (Immun-Star, Bio-Rad) and exposed to X-ray film (Kodak X-Omat Blue), scanned and densitometric analysis was performed using ImageJ software (NIH, USA).
Activation of the particular transduction pathway was presented as a ratio between the phosphorylated form and total expression of the protein (or total expression of the protein related to the expression of α-tubulin as a housekeeping gene). Mean expression in WT placebo treated animals was arbitrarily set as 1 and protein levels were expressed as fold change in relation to the mean specific protein amount in the WT placebo group.
Histology
Formalin-fixed tissue was embedded in paraffin and cut into 5 μm sections perpendicular to the long axis of the LV. Sections were stained with hematoxylin and eosin and calibrated photographs were taken under 20× magnification. Olympus Cell^D software was used to assess the cardiomyocyte cross-sectional area (CSA). A perimeter of at least 150 horizontally intersected cardiomyocytes from each animal (n = 5 per group) was marked manually and the area was returned by the software. Another set of paraffin-fixed slides (n = 5 for NaCl-treated groups and n = 8 for ISO-treated groups) was stained for fibrous tissue using the Picrosirius Red method as previously described [31]. Photographs were taken under 10× magnification from the intramyocardial portion of the left ventricle of each section and analyzed with ImageJ software using a macro-based automated analysis [32]. The area of fibrosis was expressed as the fraction of the view area occupied by collagen. Subendocardial and subepicardial regions of the LV muscle were not taken for analysis because they showed marked variability between different portions of the section.
Real-time PCR
Total RNA was isolated from left ventricular mouse myocardium of animals treated by ISO for 16 days. Directly after removal from the freezer, the material was fragmented using a hand homogenizer in a TRI Reagent Solution (Ambion). We then transferred the supernatant to a new tube and added chloroform. After mixing and incubation at room temperature homogenates were briefly centrifuged and the colorless upper aqueous phase was transferred to a new RNase-free tube. Subsequently, we added 96% ethyl alcohol in a volume corresponding to half the volume of the aqueous phase. RNA was purified using the RNeasy Kit (Qiagen). Concentration of the resulting mixture was tested for RNA concentration and purity using NanoDrop apparatus (Thermo Scientific).
Material purified from genomic DNA was collected for a total of 1 μg of RNA. Prepared RNA was then transcribed into cDNA using the commercial RT2 Profiler PCR Array System kit. The next step was to prepare the reaction mixture for real-time PCR including Master Mix, cDNA, and water. For each of the 96 wells with adsorbed primers we applied a 25 μl sample and the reaction mixture. Results of the analysis were calculated with respect to a reference gene (β-actin) amplified from the same cDNA and expressed as the difference in Ct values of individual genes for two genotypes of animals and expressed as the equation (2–ΔΔCt), where the Ct value is the threshold cycle of amplification of the studied gene. Values greater than 1 indicate greater gene expression in the animals from the IL-6 KO strain than in the WT mice. We then performed a color reaction with SYBR Green I/ROX using a 7900HT Fast Real-Time System thermocycler (Applied Biosystems).
Statistical analysis
Statistical analysis was performed using the Statistica 8.0 PL package. Data on graphs are presented using box and whiskers plots, where median and 25th–75th% percentile are marked or as mean ± standard error of the mean (SEM). Distribution of data was tested using the Kolmogorov-Smirnov test. Statistical analyses were performed using one-way analysis of variance (ANOVA) and Bonferroni’s post hoc test or Kruskal-Wallis and Dunn’s tests where applicable. A p < 0.05 was considered statistically significant.
Results
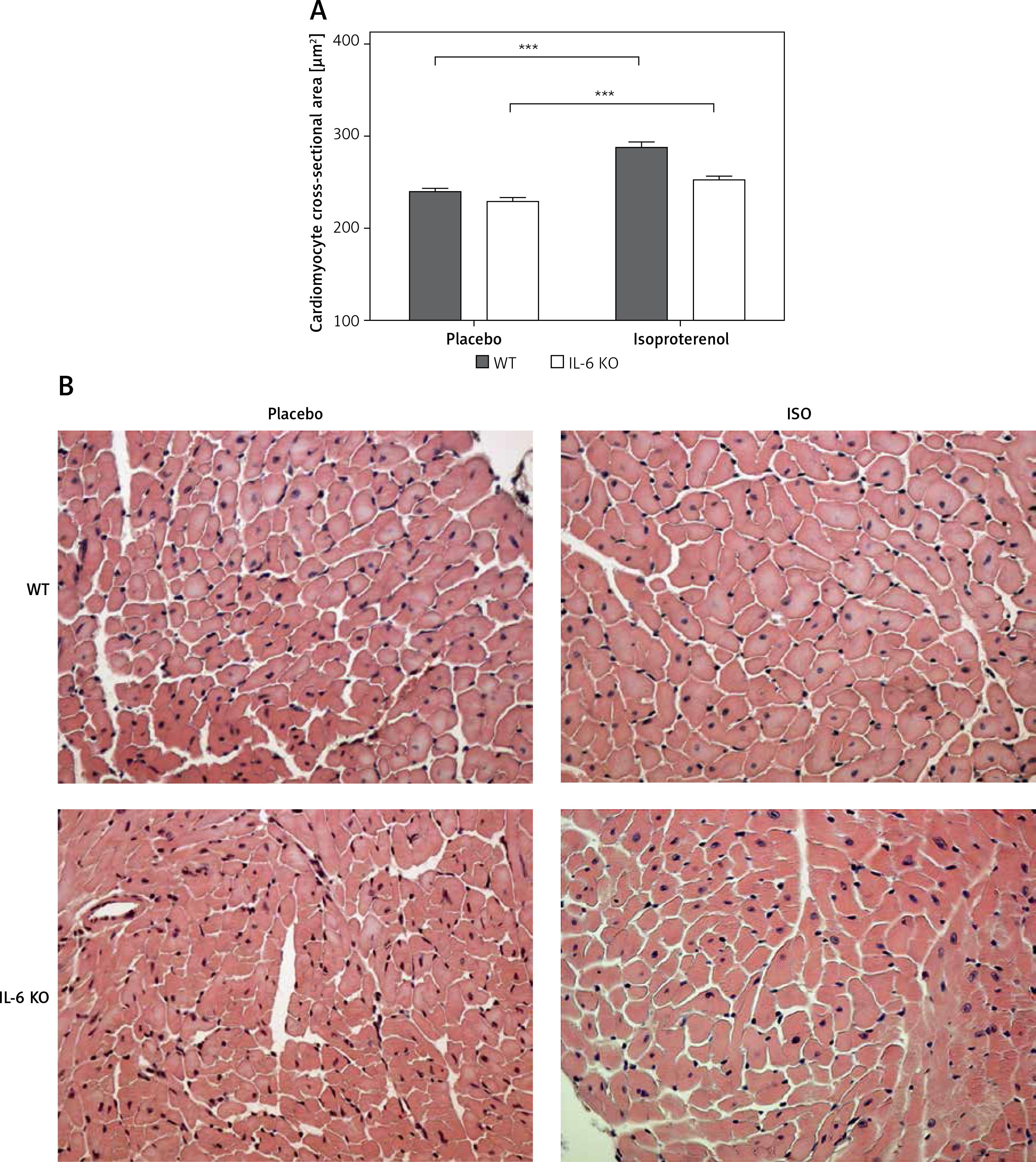
Cardiomyocyte cross-sectional area was similar in both placebo groups (WT: 239.1 ±97.6 vs. 229 ±92.6 μm2 in IL-6 KO; p = NS). ISO administration caused a significant increase in CSA in both groups: a 20% increase in WT animals (p < 0.001 vs. WT placebo) and a 10% increase in IL-6 KO animals (p < 0.001 vs. IL-6 KO), but the difference between genotypes in CSA after ISO administration was not statistically significant (Figure 1).
Figure 1
Cardiomyocyte cross-sectional area in animals treated with isoproterenol (50 mg/kg body weight) once a day for 16 days. Columns represent mean ± SEM, ***P < 0.0001. n = 5 in each group. ANOVA with Bonferroni post-hoc test. Representative H&E microphotographs are presented (magnification 20×)
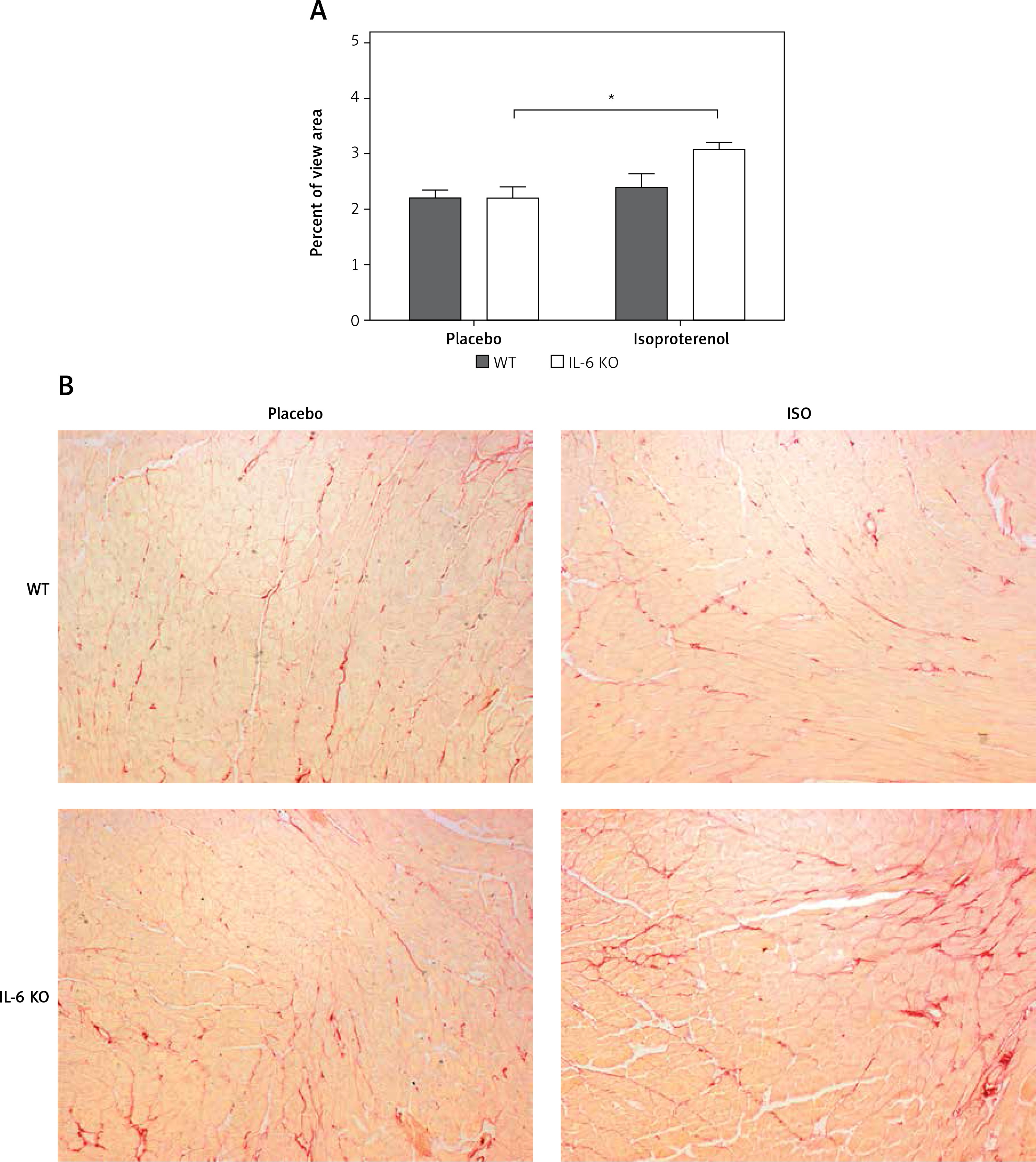
Intramyocardial fibrotic tissue occupied the same mean area of the left ventricular muscle in placebo groups (2.2 ±0.3% in WT placebo and 2.2 ±0.5% in IL-6 KO placebo group). Treatment with ISO induced a significant increase in myocardial connective tissue content in IL-6KO animals (3.1 ±0.4%, p = 0.03 vs. placebo group), while in WT mice there was no significant increase in fibrosis after ISO (2.4 ±0.5%, p = NS) (Figure 2).
Figure 2
Intramyocardial fibrosis expressed as fraction of the view area occupied by collagen in animals treated with isoproterenol (50 mg/kg body weight) once a day for 16 days. Columns represent mean ± SEM. N = 5 in placebo groups and n = 8 in ISO groups. *P = 0.03. Kruskal-Wallis test with Dunn’s post hoc analysis. Representative microphotographs showing collagen stained in red (magnification 10×)
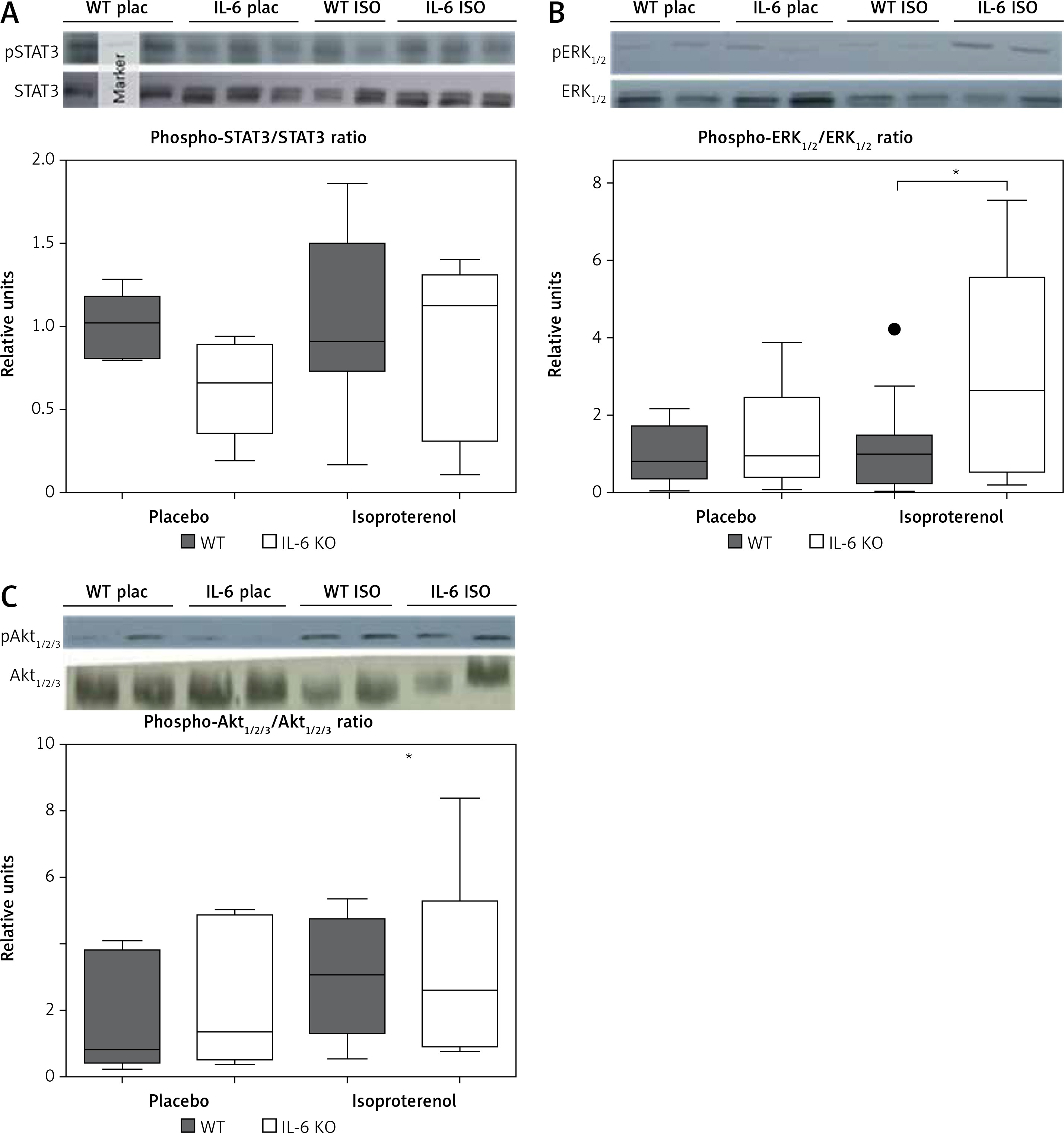
In the placebo treated animals of both genotypes baseline STAT3 protein phosphorylation was comparable. Similarly chronic ISO administration did not alter the activation of STAT3 (Figure 3 A). Phosphorylation of ERK1/2 proteins after 16 days of ISO administration in IL-6 KO animals was significantly higher than in the WT group (Figure 3 B). Phosphorylation of Akt1/2/3 proteins was not significantly elevated after ISO treatment (Figure 3 C).
Figure 3
Western blots and densitometric analysis presenting phosphorylated-to-total STAT3 proteins (A), phosphorylated-to-total ERK 1/2 (B) and Akt1/2/3 (C) kinases after 16 days of isoproterenol injections (50 mg/kg body weight ip) in left ventricular lysates of WT and IL6 KO mice. Units represent fold change in relation to the mean in WT placebo group, which is set as 1. Box represents 25th–75th percentile and the line in the middle of the box is the median. *P < 0.05. n = 6 in control groups, n = 10–11 in ISO groups. Kruskal-Wallis test with Dunn’s post hoc analysis
p38 phosphorylation was significantly increased after 16 days of ISO administration in IL-6KO mice as compared to the animals receiving saline, whereas in WT mice phosphorylation of p38 remained unchanged (Figure 4 A).
Figure 4
Western blots and densitometric analysis presenting phosphorylated-to-total c-Raf (A) and p38 (B) kinases after 16 daily isoproterenol injections (50 mg/kg body weight ip) in left ventricular lysates of WT and IL6 KO mice. Units represent fold change in relation to the mean in WT placebo group, which is set as 1. Box represents 25th–75th percentile and the line in the middle of the box is the median. *P < 0.05. N = 6 in control groups, n = 10–11 in ISO groups. Kruskal-Wallis test with Dunn’s post hoc analysis
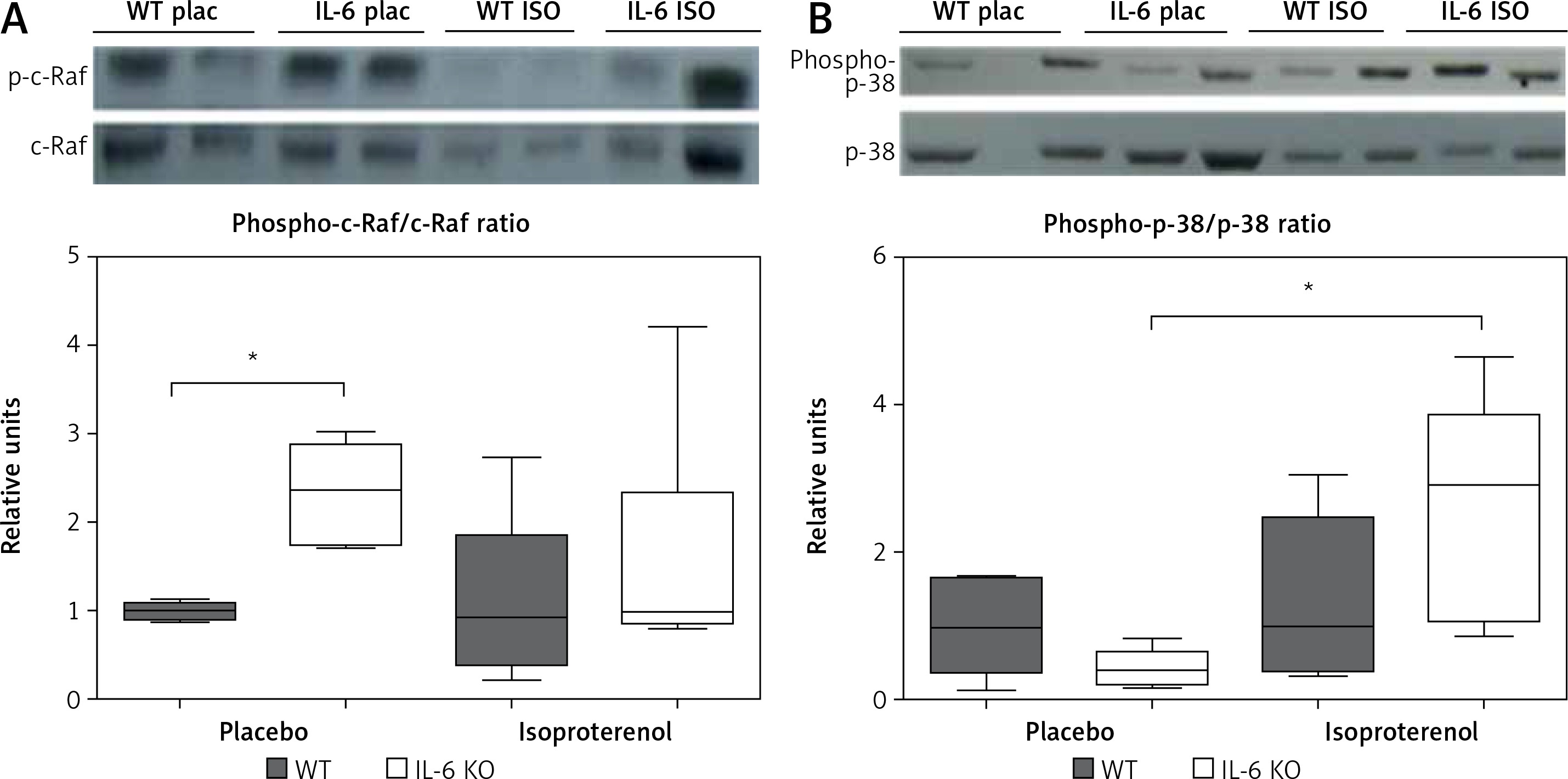
In basal conditions (placebo) c-Raf protein phosphorylation in the IL-6 KO group was significantly higher than in the WT mice. Treatment with ISO did not substantially affect phosphorylation of this protein in either genotype (Figure 4 B).
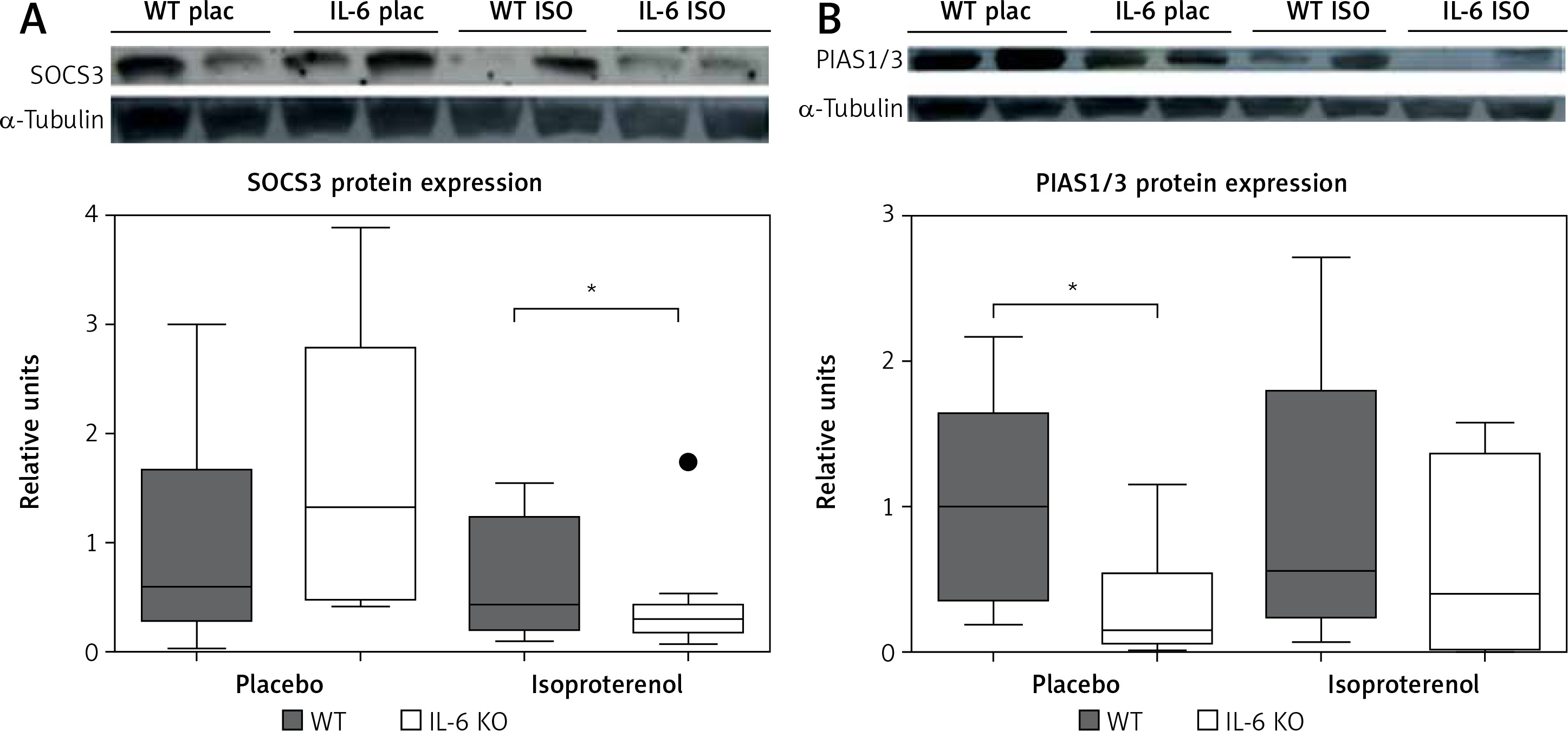
Chronic administration of ISO in the IL-6 KO group significantly reduced the expression of SOCS3 protein in comparison to the placebo group. No such effect was observed in the WT group (Figure 5 A). The amount of PIAS1/3 proteins in the IL-6 KO group receiving saline was significantly lower than in the WT group. There was no statistically significant effect of repeated use of ISO on the expression of PIAS1/3 proteins in the animals of either genotype (Figure 5 B).
Figure 5
Western blots and densitometric analysis presenting expression of SOCS3 (A) and PIAS1/3 (B) proteins normalized to the tubulin expression after 16 daily isoproterenol injections (50 mg/kg body weight ip) in left ventricular lysates of WT and IL6 KO mice. Units represent fold change in relation to the mean in WT placebo group, which is set as 1. Box represents 25th–75th percentile and the line in the middle of the box is the median. *P < 0.05. N = 6 in control groups, n = 10–11 in ISO groups. Kruskal-Wallis test with Dunn’s post hoc analysis
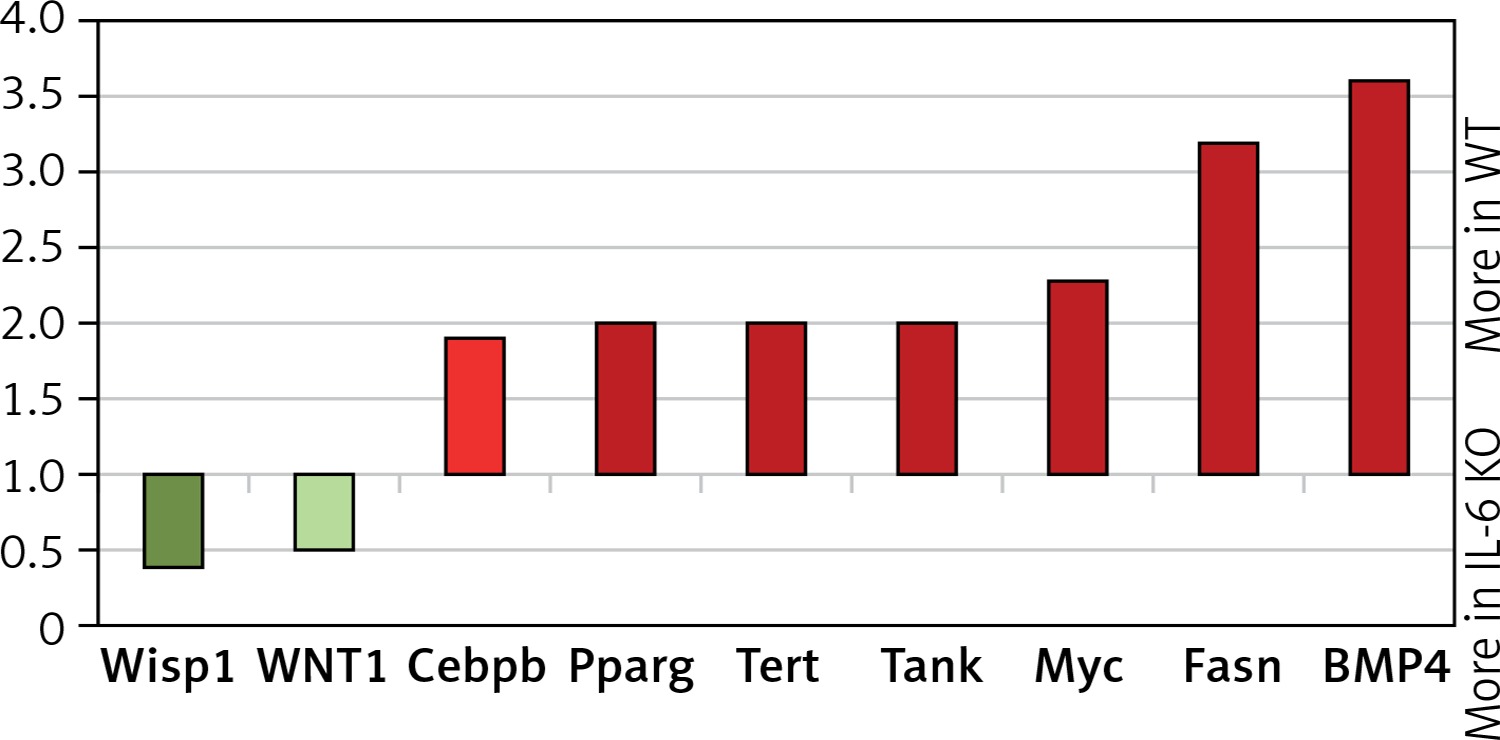
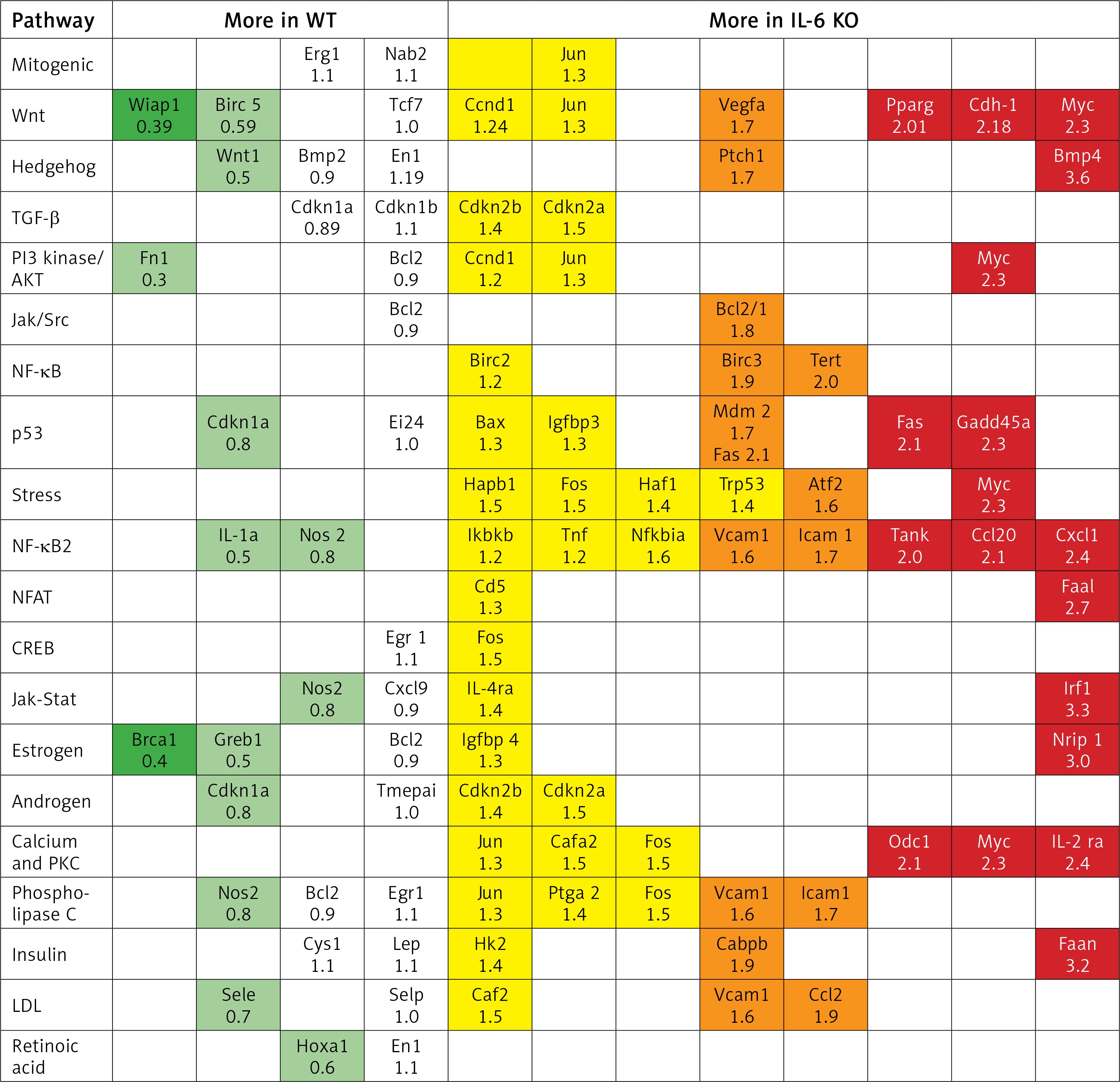
In the IL-6 KO group that received ISO for 16 days expression of genes activated by the MEK/ERK pathway was enhanced (myc-2.3x, CEBPB-1.95x, BMP4-3.6x; Fasn-3.2x; Tert-2x; Tank-2.1x), while expression of genes repressed by ERK1/2 was reduced (Wisp1, Wnt1) compared to the reference WT group (Figure 6). ISO stimulation in the IL-6 KO animals appeared to promote intracellular signaling pathways associated with NF-κB, protein kinase C (PKC) and Ca2+ (Figure 7).
Figure 6
Relative levels of transcription of genes whose activation is regulated by MEK/ERK kinase cascade pathway in the left ventricle mouse myocardium, which received ISO for 16 days assessed by real-time PCR. Expression of genes in wild-type animals adopted as 1
Wisp1 – WNT1-inducible-signaling pathway protein 1, Cebpb – CCAAT/enhancer-binding protein β, Pparg – peroxisome proliferator-activated receptor g, Tert – telomerase reverse transcriptase, Tank – TRAF-associated NF-kappaB activator, Myc – c-myc proto-oncogene, Fasn – fatty acid synthase, BMP4 – bone morphogenetic protein 4.
Discussion
Numerous previous reports demonstrate that chronic β-adrenergic stimulation, resulting from sympathetic nervous system activation, leads to cardiac remodeling and cardiovascular diseases [33, 34]. Both in vitro and in vivo studies show that cardiac remodeling in rats due to chronic adrenergic stimulation is characterized by hypertrophy of the cells and activation of the fetal gene program [35]. Intraperitoneal injections of isoproterenol in mice increased the expression of IL-6 family cytokines genes (IL-6, IL-11, oncostatin M, leukemia inhibitory factor, ciliary neurotrophic factor and cardiotrophin 1) and IL-6 level in the serum and myocardium [28].
Previous studies indicate the relationship between activation of the β-adrenergic receptor/cAMP/protein kinase A cascade and expression of the IL-6 gene [26]. It has been shown in cell cultures that an increase in the cellular concentration of cyclic AMP intensifies expression of the IL-6 gene in lung fibroblasts, while it may inhibit it in glomerular mesangial cells, monocytes/macrophages and osteoblasts [36, 37]. Stimulation of β-adrenergic receptors enhances the production of cAMP by cardiomyocytes, activating two separate intracellular signaling pathways, protein kinase A and EPAC (Exchange Protein Activated by cAMP) [38, 39], involved in IL-6 production in mouse macrophages. In cultured cardiac myocytes Epac attenuated the inhibitory effect of IL-6 on the increase of intracellular Ca2+ concentration and contractility in response to isoproterenol, most likely through inhibition of the Jak-STAT pathway via SOCS3 [40].
Zhang et al. [41] observed that infusion of ISO for 7 days, in mice with cardiac myocyte-specific deletion of STAT3, causes downregulation of transcripts for the major components of excitation-contraction coupling (mRNAs coding β1-adrenoceptor, adenyl cyclase, protein kinase A subunits, ryanodine receptor 2, and voltage-gated L-type calcium channel subunits), disengagement of calcium coupling and muscle contraction. Chronic β-adrenergic stimulation in STAT3 KO hearts results in cardiomyocyte hypertrophy, cell death and cardiac fibrosis. Phosphorylation of β1- and β2-adrenoceptors leads to activation of cardiac STAT3 through Src tyrosine kinase. Thus, cardiac STAT3 can affect β-adrenergic signaling [42]. Furthermore, STAT3 deletion in transgenic mice leads to several metabolic imbalances, including glucose transporter type 4 downregulation, resulting in incapacity of cardiac myocytes to upregulate glycolysis in response to β-adrenergic stimulation, decreased fatty acid β-oxidation and increased production of mitochondrial oxidative compounds [43].
Szabo-Fresnais et al. [26] showed that binding of circulating receptor sIL-6R with IL-6 activates protein phosphorylation of STAT3 and cellular markers of cardiac hypertrophy, whereas activation of only membrane-bound receptors leads to stimulation of other pathways via activation of kinases ERK1/2 and Akt in isolated adult rat ventricular myocytes.
Although IL-6 plays a role in cardiac hypertrophy, inhibiting the action of the cytokine by specific antibodies only partially inhibits cell hypertrophy and collagen synthesis in rat heart [44], which may indicate that the activation of intracellular pathways may depend on IL-6 as well as a number of other regulatory proteins [7]. The baseline IL-6 level is however very low in WT mice and in our previous paper we reported that left ventricular dimension and function were not different between WT and IL-6 KO mice [45]. In the presently described model, ISO induces cardiomyocyte hypertrophy in both WT and IL-6 animals, which is slightly more pronounced in the WT group, but the difference between genotypes is insignificant.
Interleukin 6 plays an important role in driving fibrosis in unresolved chronic inflammation [46]. Infusion of IL-6 lasting 1 week caused a significant increase of collagen deposition in rats [8]. Our data show comparable content of collagen in the myocardium of both WT and IL-6 KO mice in basal conditions, while treatment with ISO stimulates interstitial fibrosis of the myocardium only in IL-6 KO animals, which may be the consequence of augmented activation of MAP kinase pathways. The inconsistencies between stimulation protocols showing clear IL-6-dependent fibrotic response and our study may result from differences in IL-6 concentration: In stimulation studies usually supraphysiological concentrations are used and the intact mice of WT that were used in this project have a very low basal level of the cytokine, which may explain the lack of pronounced effects of IL-6 knock-out on collagen deposition in our results. Another explanation for the lack of clear phenotypic differences between genotypes could be compensatory dysregulation of other pathways in IL-6 KO animals. IL-6 is one of the family of cytokines able to evoke similar cellular responses. For example, cardiotrophin-1 has been shown to regulate hypertrophy and fibrosis in coculture experiments using adult mouse cardiac myocytes and fibroblasts [47].
Previous studies have shown that the lack of IL-6 does not affect the activation of STAT3 and ERK1/2 after a short bout of adrenergic stimulation in mouse [29]. In the present study chronic ISO administration caused an increase of phosphorylation of ERK kinases while having no effect on STAT3 activation. We found that deficiency of IL-6 during chronic administration of ISO intensifies the gene expression usually promoted by phosphorylated ERK proteins, such as Cebp-β, PPAR-γ, Tert, Tank, Myc, Fasn or BMP4. These results may indicate the activation of successive stages of the ERK cascade. It is important to emphasize that our results do not present the effect of ISO injection, but reflect the equilibrium in the myocardium subjected to chronic adrenergic stress. Lack of IL-6 may induce compensatory mechanisms to maintain the allostasis under isoproterenol stimulation. In a model of cardiac pressure overload in mice with IL-6 knockout decreased IL-11 expression was proposed to compensate for lack of IL-6 [48].
The intracellular signaling pathways of IL-6 are negatively regulated by several proteins, including SOCS. The gene responsible for the SOCS3 expression belongs to the early response gene family regulated by factors such as IL-6 and LIF, IL-1 or leptin [49], whose levels increase in the absence of IL-6. SOCS3 competes with SHP2 for the phosphorylated Y757 docking site and thereby reduces gp130-dependent ERK activation mediated by SHP2 [23]. We demonstrated that in the absence of IL-6, chronic β-adrenergic stimulation reduces the expression of SOCS3 protein, which may be, at least partially, responsible for increased activity of ERK1/2 and p38 kinases. However, the lack of concomitant increase of c-Raf phosphorylation, which could be expected, may suggest involvement of other mechanisms in addition to of downregulation of SOCS3. These effects were not observed in WT animals and suggest a suppressive effect of IL-6 on MAPK activation. In a study conducted on a model of transgenic animals with cardiac-specific SOCS3 knockout, Yajima et al. observed development of contractile dysfunction and ventricular arrhythmias with only minimal changes in myocardial histology but increased activation of gp130 downstream signaling targets STAT3, ERK1/2, Akt and p38 [50]. In another study from the same group, deletion of SOCS3 prevented remodeling of the heart after myocardial infarction through inhibition of apoptosis and fibrosis, as well as augmentation of antioxidant mechanisms, which were related to enhanced activation of STAT3, AKT, and extracellular signal-regulated kinase (ERK)-1/2 [51].
SOCS3 protein expression is induced by a number of cytokines, while PIAS1/3 protein is expressed in a constitutive manner. The concentration of PIAS1/3 was not affected by β-adrenergic stimulation in our data, but it was lower at baseline in IL-6 KO mice.
In conclusion, IL-6 attenuates activation of MAP kinases induced by chronic isoproterenol administration in the mouse myocardium, which may be related to maintenance of SOCS3 expression by IL-6 and protects the heart from fibrosis. β-adrenergic stimulation induces cardiomyocyte hypertrophy, which is not altered by IL-6.