Introduction
Hepatocyte nuclear factor 4α (HNF-4α) is an orphan member of the nuclear receptor family of ligand-activated transcription factors that are activated by steroid hormones. The HNF-4α gene is evolutionarily conserved across multicellular organisms and encodes a protein that regulates transcription through homodimer DNA binding to its target genes [1, 2]. Research in several mammals shows that the HNF-4α protein is a nuclear DNA-binding phosphoprotein that acts as a transcriptional activator or repressor of several target genes [2–5]. HNF-4α is involved in the regulation of 40% of hepatic genes and 11% of pancreatic islet genes [6]. This transcription factor is necessary for development and differentiation that influence a wide variety of cell types and tissues with roles in gluconeogenesis [7], fatty acid metabolism [8], drug metabolism [9], hepatocyte differentiation [10] and blood coagulation [11]. HNF-4α was first detected in crude liver extracts by Sladek et al. in 1990 and its gene resides on chromosome 20 [12]. Phylogenetic research suggests that HNF-4α evolved from an explosive burst of gene duplications that occurred about 541 million years ago [13].
HNF-4α in development and normal physiology
A comprehensive explanation of HNF-4α’s roles in development and physiology is beyond the scope of this article. Therefore, here we briefly review the fundamental features. HNF-4α plays essential roles in cross-regulatory networks that are essential in the development of the pancreas, liver, and kidneys during the embryonic stage. In typical development, HNF-4α is highly expressed initially in the embryonic endothelium, where it plays a critical role in hepatogenesis and the formation of hepatoblasts [14–16]. Additionally, HNF-4α is highly expressed in the embryonic digestive system and neural crest cells during their development stage [17]. HNF-4α expression decreases with age. However, its expression is not restricted to development as it continues to regulate gene expression in adult hepatocytes and pancreatic islets [18]. During the embryonic development of mice, HNF-4α is initially expressed in the endodermal cells, particularly the yolk sac, in the hindgut and liver diverticulum. Later on, HNF4 isoforms were detected in the developing kidneys, stomach, and pancreas. HNF-4α is essential for organogenesis, in which it functions as a development regulator [17]. Recently, several studies have shown that HNF-4α transcripts regulate each other in a synergistic pattern and regulate the development, integrity, viability and functions of the embryo and numerous adult tissues. With these observations, hepatocyte-specific HNF-4α knockout in mice leads to hepatomegaly and an increase in intrahepatic fat that is associated with embryonic lethality [19, 20]. HNF-4α has also been shown to be involved in intestinal inflammation interactions, where it protects the mucosal layer of the intestines from prolonged and extreme inflammatory conditions [21]. Inhibition of HNF-4α in non-transformed immortalized human hepatocytes leads to activation of the inflammatory IL-6/STAT3 pathway, which suggests that HNF-4α acts as a repressor of STAT3 [21, 22]. HNF-4α has also been identified as a transcriptional activator of factor VII and, therefore, plays a significant role in blood coagulation. A mutation in the HNF-4α binding region of the factor VII promoter resulted in limited interaction between HNF-4α and its binding site, which reduced the promoter activity and led to severe factor VII deficiency [23]. Moreover, HNF-4α is essential for the kidneys as it is expressed in the proximal tubules. In the adult rat nephrons, it plays an important role in the formation of developed proximal convoluted tubules. On the other hand, the loss of HNF-4α caused a reduction in the expression of multiple genes that are expressed in the proximal tubules. The aberrant expression of HNF-4α in the proximal tubules promotes defective development and accumulation of calcium in the kidneys [24]. In the embryonic pancreas of mice, HNF-4α has been described as a major regulator of several transcriptional interactions that are essential for the differentiation and specification of the pancreatic cells. HNF-4α is constitutively expressed in the pancreatic exocrine glandular cells, where it controls the function of mature β-cells. Mutations in HNF-4α and HNF1α genes lead to decreased insulin secretion, which causes diabetes [25].
Structure of HNF-4α protein
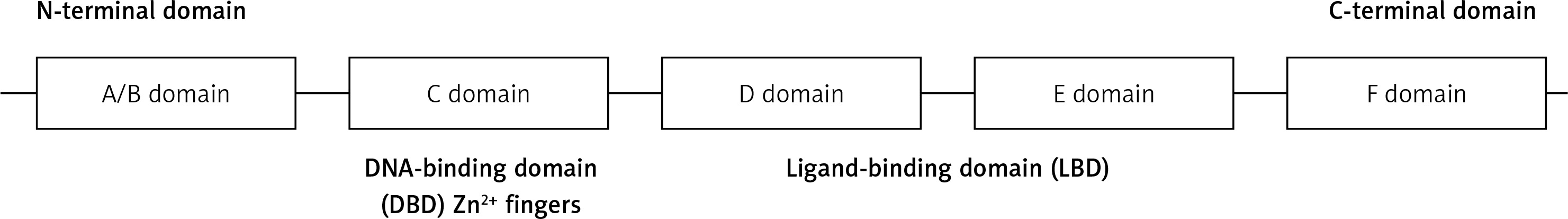
Full-length HNF-4α is a 465 amino-acid 78-kDa transcription factor [26]. What identifies the nuclear receptor superfamily is a specific DNA-binding domain (DBD) [27]. HNF-4α is characterized by five working domains (A/B, C, D, E, and F domains). DBD is located in the C domain, which is a 60–90 amino-acid domain that consists of two zinc finger modules followed by a C-terminal extension (Figure 1). The core recognition motif in the DBD consists of a six-base pair sequence, which is responsible for target recognition. This motif is conserved for all members of the nuclear receptor superfamily, whereas other structures vary to serve unique receptor-specific binding sites. The transcriptional response elements within HNF-4α target genes result from three different manners of DNA binding: monomers, homodimers, or heterodimers. The ligand-binding domain (LBD), which is located in the D/E domain, is composed of approximately 200 aa. LBD plays several roles, such as protein dimerization, transactivation, and ligand binding. The formation of homodimers or heterodimers in a solution is essential for protein-protein interactions, and it is controlled by LBD [26, 27]. HNF-4α phosphorylation by ERK1 revealed several phosphorylation sites located in the DBD, LBD, and the hinge and the C-terminus of HNF-4α. The phosphorylation of HNF-4α at multiple sites, such as serine and threonine residues, leads to the inhibition of its transcriptional activation capability. The mechanism of HNF-4α inhibition by the activated ERK pathway could also be achieved by cytokines, oxidative stress and growth hormones. This inhibition leads to reduced HNF-4α transactivation abilities and, consequently, the downregulation of its target genes [27].
Involvement of HNF-4α in haematological disorders
Over the last decade, HNF-4α has been increasingly implicated in the aetiology of different haematological conditions. In 2015, a paper published by Shunsuke et al. reported that HNF-4α regulates both iron metabolism and transferrin receptor 2 (Tfr2) and, therefore, plays a significant role in iron deficiency anaemia. This study showed that the elimination of HNF-4α in liver-specific HNF-4α-null mice lowered the serum iron levels but did not cause significant variations in the red blood cell (RBC) count, haemoglobin concentration and haematocrit. This means there was no evidence of iron deficiency anaemia in these mice. Nevertheless, there was a remarkable reduction in the levels of hepatic transferrin mRNA and serum transferrin protein in HNF-4α-null mice. This low expression resulted from the limited transcriptional activation of transferrin by HNF-4α [28]. Another study reported that in mice carrying a mutation in the transferrin gene, a hypotransferrinaemic status developed, resulting in severe iron deficiency anaemia that killed the mice. However, direct injection of serum or purified transferrin rescued these mice [29]. Hence, hypoferremia cannot be fully attributed to the elimination of HNF-4α. Therefore, the reduction in the serum transferrin protein that resulted from the lack of HNF-4α was not sufficient to induce iron deficiency anaemia [29]. A recent study has correlated the production of hepatic transferrin with serum transferrin. They also suggested that HNF-4α regulates hepatic transferrin production and HNF-4α signalling activity could be reflected through the serological testing of transferrin levels [30]. Additionally, the role of HNF-4α has been reported to be associated with erythropoiesis in humans [31].
Fetal haemoglobin (HbF) levels are high in cases of sickle cell anaemia. A study evaluated HbF in erythroid progenitor cells, human umbilical cord blood-derived erythroid progenitor-2 (hudep-2) aiming to understand its regulation. Following the induction of HbF production by treating the cells with the antineoplastic drug decitabine – a ‘hypomethylating agent’ – they observed an elevation in HNF-4α levels. Accordingly, in this study HNF-4α was chosen as a target for knockdown, which increased HbF levels. Therefore, the findings of this study suggest that HNF-4α might regulate the transcription of γ-globin, which is a component of HbF [32].
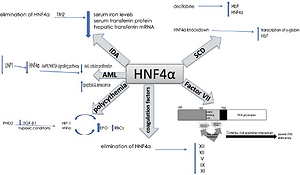
Moreover, in acute myelogenous leukaemia (AML), HNF-4α was found to be a part of a regulatory mechanism that is essential for AML cell survival. Shi et al. reported that HNF-4α is regulated by LncRNA in non-homologous end-joining pathway 1 (LINP1) protein, which is overexpressed in AML cells and accelerates disease progression. The survival and glucose metabolism of AML cells depend on HNF-4α and its downstream AMPK/WNT5A signalling pathway. LINP1 inhibition leads to HNF-4α downregulation and, subsequently, reduced AML cellular proliferation and induced apoptosis and senescence. These findings suggest that HNF-4α could be used as a biomarker and a target for therapy in AML [33]. Another study that aimed to compare the patterns of kinetic changes in gene expression between sensitive and resistant multiple myeloma (MM) patients found that HNF-4α is one of the activated genes in the resistant patients. Exploration of the association of HNF-4α with the therapeutic resistance of MM would be an interesting topic for future research [34] (Figure 2).
Figure 2
Summary of the role of HNF-4a in haematological disorders. In iron deficiency anaemia, HNF-4a has been shown to lower serum iron levels, serum transferrin protein and hepatic transferrin mRNA. In sickle cell anaemia, HNF-4a siRNA knockdown and chemical induction both enhanced the transcription of γ-globin and HbF levels. HNF-4a is regulated by LINP1in acute myelogenous leukaemia, where the cell survival depends on HNF-4a and its downstream AMPK/WNT5A signalling pathway. HNF-4a increases EPO expression and erythrocytosis in polycythaemia, and EBO is regulated by HNF-4a, which is maintained by TGF-β. Under hypoxic conditions, both HNF-4a and HIF-1 regulate EBO production. The elimination of HNF-4a results in reduced expression of XIII, XII, V, IX and XI coagulation factors. HNF-4a binds directly to its promoter sequence at the −33 to −50 bp region on the FVII gene. This binding is disrupted by two-point mutations at the promoter region, thymine to cytosine at –60 bp and arginine replacement with histidine at position 348. Consequently, severe FVII deficiency occurs
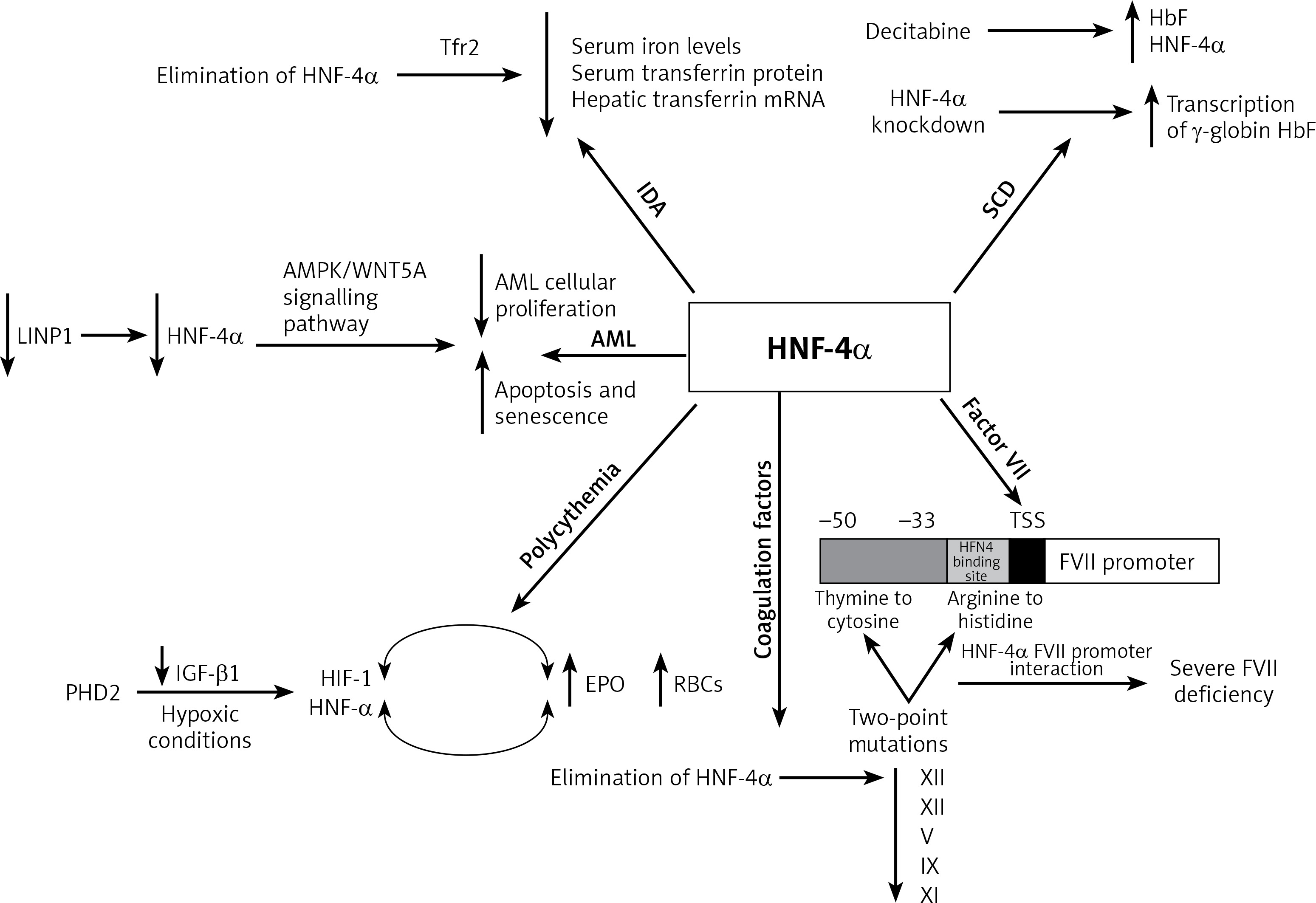
Increased RBC count (polycythaemia) is another haematological condition where HNF-4α expression was apparent. Polycythaemia and its related erythrocytosis increase the risk of blood clots and hyperviscosity. Erythrocytosis is one of the main disorders in hepatocellular carcinoma (HCC) which results from the overexpression of erythropoietin (EPO), which is a hormone produced by the kidneys to stimulate the production of RBCs. HNF-4α transcriptionally regulates EBO. Under hypoxic conditions, HNF-4α and another DNA-binding transcription factor known as hypoxia-inducible factor 1 (HIF-1) regulate EPO expression. In HepG2, a human hepatocellular carcinoma cell line that secretes EPO, HNF-4α was found to be maintained by prolyl-4-hydroxylase 2 (PHD2) induced suppression of the TGF-β1 pathway. Thus, HNF-4α enhances the expression of EPO and thereby promotes erythrocytosis [35–37].
Furthermore, HNF-4α is required for optimal gene expression of several blood coagulation factors. For instance, low expression of XIII, XII, V, IX and XI coagulation factors was observed in HNF-4α-null mice. HNF-4α binds directly to its promoter sequence on the genes that encode these coagulation factors and activate their transcription [38–40]. Compared to the wild-type mice, there was a slight reduction in IX and no difference in VII, VIII, or X levels of expression in HNF-4α-null mice. On the other hand, the complete absence of HNF-4α resulted in low expression of XIII and XII. A recent study observed elevation of HNF-4α expression that had been induced by histone H4 and DNA. Overexpression in the mRNA of several coagulation factors was partially triggered by the increase in HNF-4α expression [41]. This suggests that HNF-4α transcriptional activity is essential for the gene expression of XIII and XII among other coagulation factors.
Factor VII (FVII) is another coagulation factor that is transcriptionally regulated by HNF-4α. Stauffer et al. identified the −33 to −50 bp region upstream of the transcription start site within the FVII promoter as the HNF-4 binding site [39]. Two-point mutations at the promoter region of the HNF-4α binding site contributed to a severe FVII deficiency. The first point mutation is the transformation of thymine to cytosine at –60 bp. This mutation abolishes efficient HNF-4α-FVII promoter interaction and leads to severe FVII deficiency. Arginine replacement with histidine at position 348 was identified as the second point mutation in the FVII gene promoter region. HNF-4α transcriptional regulation of factor VII is highly significant since it plays an important role in the coagulation cascade. The activation of IX and X depends on the FVII-tissue factor interaction. Activated factor X transforms fibrinogen to fibrin, generating thrombin, which is essential for stabilizing the initial haemostatic plug [38, 39, 42, 43] (Figure 2).
Potential therapeutic role of HNF-4α
The several findings explained in the previous sections associate HNF-4α with numerous aspects of haematological disorders. The enormous body of evidence suggests that the apparent expression of HNF-4α does contribute to worse clinical outcomes and is involved in the regulation of signalling pathways that are dysregulated. HNF-4α is involved in several biological processes that are relevant to iron metabolism, HbF levels, AML cell survival, erythrocytosis and the expression of coagulation factors.
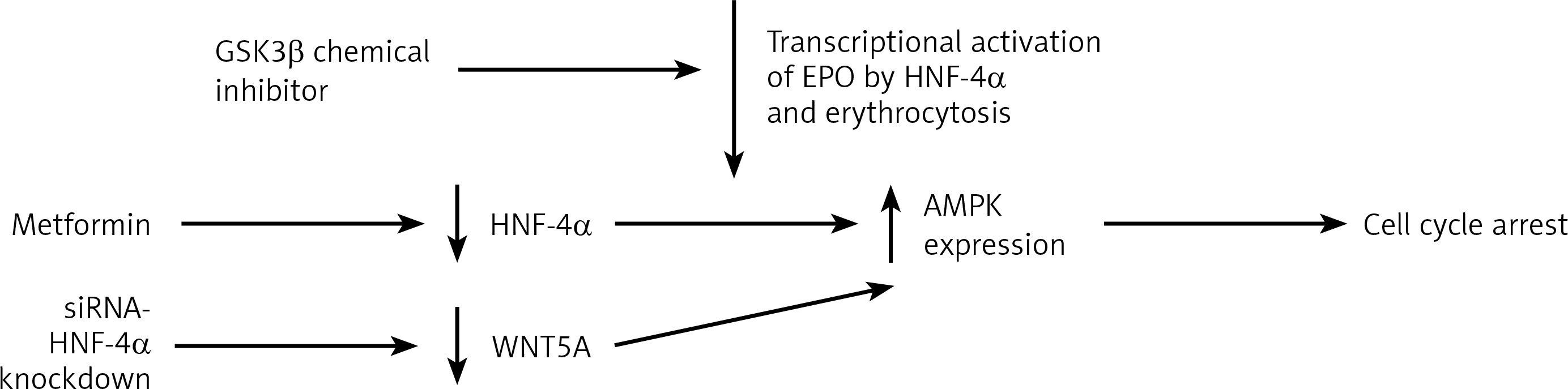
Having discussed the association of HNF-4α with haematological conditions, HNF-4α as a potential target for therapies comes next in this review. For instance, the renewal of HNF-4α functions has been proposed as a potential therapeutic strategy in HCC [44]. Due to the involvement of HNF-4α in polycythaemia, HNF-4α and its co-factors constitute a target for novel therapies. As previously described, the apparent expression of HNF-4α promotes erythrocytosis, which is one of the complications associated with hepatocellular carcinoma (HCC). The DNA binding activity of HNF-4α is maintained by the suppression of TGFβ by PHD2. This enabled HNF-4α to continue activating EPO expression, which enhances erythrocytosis [37]. TGFβ was found to weaken HNF-4α DNA binding activity on HNF-4α target genes [45]. This TGFβ dominance over HNF-4α occurs because of the inactivation of glycogen synthase kinase 3 β (GSK3β). The post-translational modifications profile of HNF-4α was found to be moderated by the activity of GSK3β. This moderation was the result of the chemical inhibition of the inhibitor BIO by GSK3β. This led to the loss of the negative charge on HNF-4α, which impaired its DNA binding activity [45]. Therefore, the GSK3β inhibitor could be used to diminish the transcriptional activation of EPO by HNF-4α and lower the HCC associated erythrocytosis. In general, targeting HNF-4α demonstrated anti-tumour activities in gastric cancer [46], prostate cancer [47] and colorectal cancer [48]. Targeting HNF-4α caused AMPK to link to the Wnt signalling pathway in the cases of gastric cancer (GC). Applying metformin, an HNF-4α antagonist, on GC cell lines reduced the expression of HNF-4α by increasing AMPK expression and induced cell cycle arrest. WNT5A has been reported to be an HNF-4α target gene where the downregulation of WNT5A occurred as a result of siRNA-mediated knockdown of HNF-4α. This indicates that HNF-4α antagonists exerted antitumor activities in GC [46]. Wnt signalling is essential for several regulatory functions in AML and other leukaemias [49] (Figure 3). In AML, cell survival depends on HNF-4α and its downstream AMPK/WNT5A signalling pathway. Similar to GC, the AMPK- HNF-4α-WNT5A signalling pathway represents a potential therapeutic target in AML. These findings suggest that introducing a drug combination that incorporates HNF-4α antagonists in the AML course of therapy could be a promising development. Overall, targeting HNF-4α is another avenue for the potential treatment of several haematological conditions.
Figure 3
HNF-4a as a target of therapy. Targeting HNF-4a through the downregulation of protein level to restrain the transcriptional activity of hyperactivated HNF-4a. Targeting the transcription of HNF-4a by GSK3β inhibitor to loosen the negative charges of HNF-4a and weaken its DNA binding activity to reduce the transcriptional activation of EPO by HNF-4a. The inhibition of HNF-4α by applying metformin increases AMPK expression and induces cell cycle arrest. Another way to target HNF-4a is by siRNA-mediated HNF-4α knockdown, which results in WNT5A downregulation and increases AMPK expression, which also induces cell cycle arrest
Conclusions and future perspectives
The role of HNF-4α in several haematological conditions has been well established over the last decade. The ability of HNF-4α to regulate the transcription of a broad network of genes involved in growth, differentiation, and epigenetic control indicates its physiological potential. To associate a particular gene with a specific type of haematological disorder is extremely unusual in the field of pathology and cancer research. This review has focused on haematological conditions. However, HNF-4α is also implicated in a wide range of malignancies and hereditary disorders. It is rational to anticipate that HNF-4α will turn out to be implicated in numerous other types of metabolic disorders. The use of chemical inhibitors to disrupt the abilities of HNF-4α to interact with other proteins and cofactors or inhibit its DNA binding capacity to its target genes is yet to be studied. More research is needed before the role of HNF-4α in haematological disorders can be fully comprehended. There is no doubt that HNF-4α-based prognostic markers and therapeutic interventions will offer new opportunities in the clinical interventions of haematological disorders.