
A 69-year-old male patient was admitted to our department on May 27, 2025, due to persistent abdominal pain accompanied by abdominal distension, nausea, and acid reflux for 2 months. He had no history of viral hepatitis or alcohol abuse. Before our admission, on April 30, he underwent contrast-enhanced computed tomography (CT) scans at another hospital, showing small nodules in the liver as well as portal vein thrombosis (PVT) accompanied by cavernous transformation of the portal vein (CTPV). A slightly low-density nodule with a size of approximately 2.1 cm × 1.7 cm × 1.7 cm was visible under the capsule of the S8 segment of the liver. Similarly, another slightly low-density nodule with a size of approximately 2.5 cm × 2.7 cm × 2.2 cm was also observed in the S2 segment of the liver. On May 13, he underwent laparoscopic radiofrequency ablation for hepatic nodules at that hospital. However, no pathological biopsy was conducted; thus, the specification and grading of the nodules remain undetermined. After that, abdominal pain still persisted, but his abdomen was soft without tenderness, rebound, or muscular tension on physical examinations.
On May 28, laboratory tests showed that D-dimer was 19.58 mg/l (reference range: 0.00–0.55 mg/l), fibrinogen degradation products (FDP) was 60.92 µg/ml (reference range: 0.0–5.0 µg/ml), white blood cell count was 6.2 × 109/l, red blood cell count was 4.47 × 1012/l, hemoglobin level was 134 g/l, percentage of basophilic granulocytes was 0.5%, platelet count was 87 × 109/l (reference range: 125–350 × 109/l), haematocrit was 0.402 l/l, lactate dehydrogenase level was 222 u/l, serum direct bilirubin was 6.9 µmol/l, and indirect bilirubin was 8.7 µmol/l. Abdominal contrast-enhanced CT scans indicated portal hypertension, esophagogastric varices, and occlusive thrombosis in the portal vein (Figure 1). Subsequently, low-molecular-weight heparin sodium (LMWH) was administered subcutaneously at a dose of 5000 IU every 12 h.
On May 30, his abdominal symptoms improved. Laboratory tests showed that D-dimer was 16.53 mg/l, FDP was 38.36 µg/ml, antithrombin III (ATIII) was 64.2% (reference range: 75–125%), and homocysteine (Hcy) was 20.76 µmol/l (reference range: 0–20 µmol/l). Plasma protein S activity, plasma protein C activity, and antiphospholipid syndrome antibody were within their reference ranges. Homozygous 4G/4G mutation in the plasminogen activator inhibitor type 1 (PAI-1) gene and homozygous C677T mutation in the methylenetetrahydrofolate reductase (MTHFR) were identified.
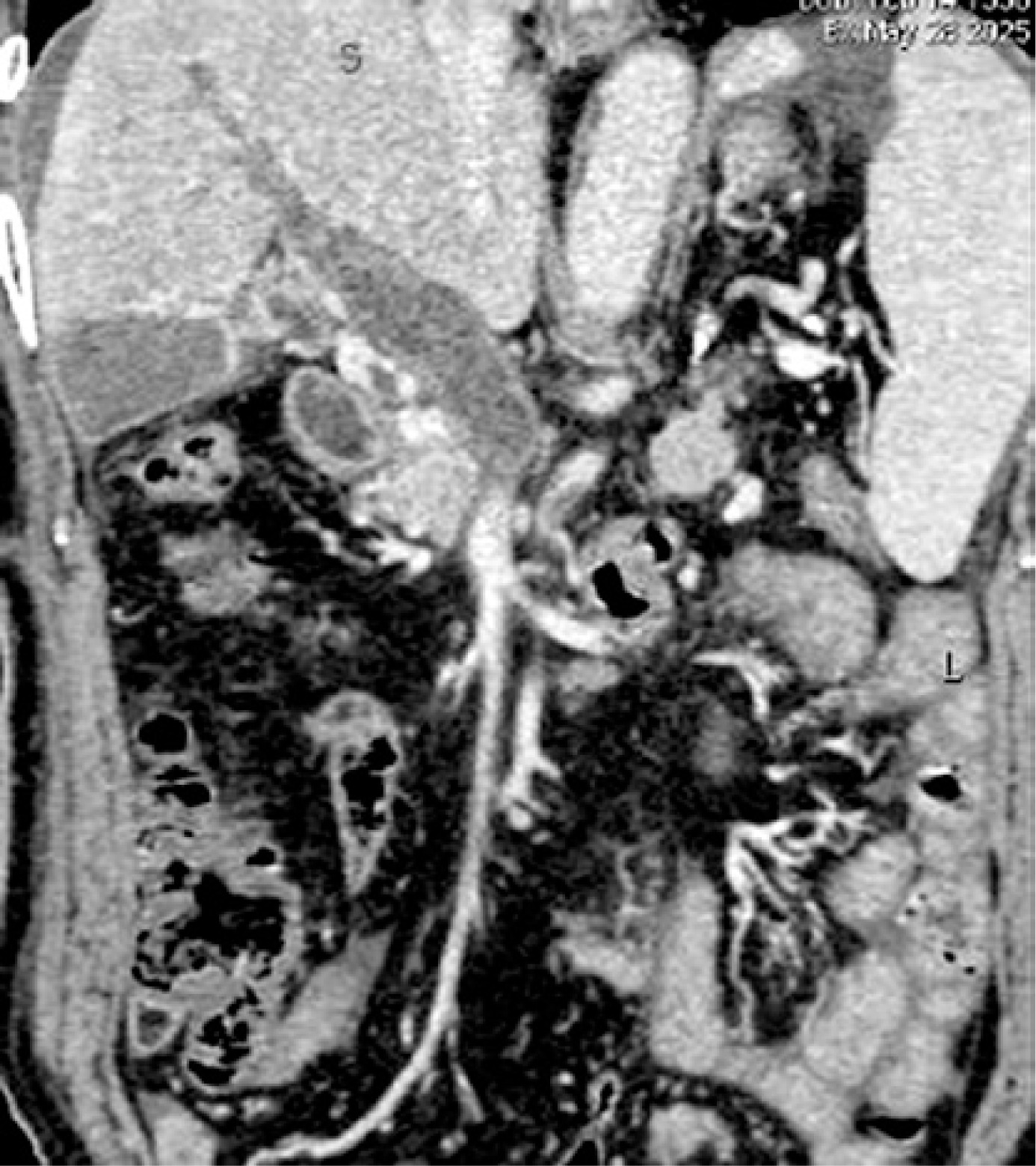
On June 7, CT angiography showed that the portal vein remained occluded. On June 11, the patient’s abdominal pain was significantly alleviated. He was discharged with oral rivaroxaban.
On June 27, laboratory tests showed that D-dimer was 2.45 mg/l and FDP was 8.18 µg/ml. CT angiography still demonstrated persistent occlusion of the portal vein and its branches, accompanied by the development of collateral vessels. He was discharged on long-term oral rivaroxaban.
PVT is rare in individuals without liver cirrhosis, but can cause lethal complications [1]. PVT is mainly attributed to Virchow’s triad [2]. Hypercoagulability can be hereditary and acquired. The former mainly includes mutations in factor V Leiden and prothrombin gene G20210A and deficiencies in anticoagulation proteins (ATIII, protein S, and protein C), while the latter mainly includes malignant tumors, myeloproliferative neoplasms, especially JAK2V617F mutation, and antiphospholipid syndrome. In addition, abdominal surgery and infections may contribute to vascular endothelial injury or reduced blood flow velocity, thereby promoting thrombosis [3]. In our case, laboratory tests showed no mutations in factor V Leiden or prothrombin gene G20210A, and the levels of anticoagulation proteins within their reference ranges. Unfortunately, the JAK2V617F gene mutation was not detected in this case. Notably, there was no abnormality in any hematologic parameters potentially associated with myeloproliferative neoplasms. Additionally, the findings from abdominal contrast-enhanced CT scans did not indicate the presence of malignant tumors. Thus, the possibility of tumor-associated thrombosis or tumor thrombosis should be excluded. Regardless, the patient had homozygous mutations in both the MTHFR C677T gene and the PAI-1 gene.
PAI-1 is the main inhibitor of tissue-type plasminogen activator and urokinase-type plasminogen activator, and its overexpression can disrupt the fibrinolytic system and increase the risk of thromboembolic events [4]. PAI-1 (4G/4G) genotype is significantly associated with venous thrombosis [5]. Zhang et al. suggested that PAI-1 (4G/5G) gene polymorphism might be a potential biomarker for venous thrombosis [6]. In our case, the patient had a homozygous mutation in the PAI-1 gene, which might promote the development of PVT.
MTHFR is a critical enzyme in the metabolism of Hcy, playing a role in maintaining normal plasma Hcy concentrations. Functional deficiency of MTHFR or reduced enzymatic activity may disrupt Hcy metabolism, leading to hyperhomocysteinemia, which is an independent risk factor for thrombotic events [7]. Homozygous or heterozygous mutations in the MTHFR gene are associated with an increased risk of venous thrombosis [8–11]. Furthermore, Ames et al. found that individuals with homozygous MTHFR C677T genotype developed PVT about 20 years earlier than wild-type carriers [12]. Multiple meta-analyses consistently suggest that MTHFR gene polymorphism may potentially increase the risk of venous thrombosis, particularly in Asian populations [13, 14]. Our case had a homozygous mutation of the MTHFR gene, providing additional evidence regarding genetic predisposition to thrombosis.
Taken together, PAI-1 4G/5G and MTHFR C677T polymorphisms play a role in the development of venous thrombosis. However, the combined effect of these genetic polymorphisms on venous thrombosis risk remains unclear. A Chinese lung cancer cohort study suggested that the combined mutations may further increase the risk of venous thrombosis, whereas an Iranian case-control study revealed no statistically significant synergistic effect [15, 16]. Therefore, the combined mutations may only moderately increase the risk of thrombosis. Beyond this, the presence of hepatic nodules or the decrease in platelet count may indicate other undetected potential prothrombotic risk factors in this patient.
Currently, the treatment selection of PVT is made in a case-by-case manner. Indeed, vascular interventional therapy is often technically difficult in extensive portal venous system thrombosis with CTPV [17]. Early initiation of anticoagulation can facilitate partial or complete recanalization of acute PVT, and prevent the development of intestinal ischemia and chronic PVT [18, 19]. Low-molecular-weight heparin is the preferred choice of anticoagulants [20, 21]. In our case, following the initiation of anticoagulation, the patient’s abdominal pain gradually improved. This may be because PVT did not worsen, and intestinal congestion was alleviated. Failure to achieve portal vein recanalization may be related to delayed initiation of anticoagulation in our case.