Current issue
Archive
Manuscripts accepted
About the Journal
Editorial office
Editorial board
Section Editors
Abstracting and indexing
Subscription
Contact
Ethical standards and procedures
Most read articles
Instructions for authors
Article Processing Charge (APC)
Regulations of paying article processing charge (APC)
HYPERTENSION / RESEARCH PAPER
Bioinformatics analysis of the differentially co-expressed genes and immune cell infiltration features associated with pulmonary arterial hypertension
1
Department of Cardiology, Affiliated Hospital of Nantong University, Nantong 226001, China
2
Department of Cardiology, The Fifth Hospital of Xiamen, Xiamen, 361101, China
Submission date: 2021-06-09
Final revision date: 2021-08-26
Acceptance date: 2021-09-09
Online publication date: 2021-09-17
Corresponding author
Xiaofei Li
Department of Cardiology, Affiliated Hospital of Nantong University, Nantong 226001, China
Department of Cardiology, Affiliated Hospital of Nantong University, Nantong 226001, China
KEYWORDS
pulmonary arterial hypertensiondifferentially co-expressed genesweighted gene co-expression network analysisfunctional enrichment analysisimmune infiltrationcell type identification by estimating relative subsets of RNA transcripts
TOPICS
ABSTRACT
Introduction:
Gene expression and immune cell infiltration are essential in the etiopathogenesis of pulmonary arterial hypertension (PAH). Our study attempted to identify novel differentially co-expressed genes and to investigate the features of immune cell infiltration in PAH.
Material and methods:
The GSE113439 and GSE117261 datasets were acquired from the Gene Expression Omnibus (GEO) database. The differentially expressed genes (DEGs) between the PAH and control groups were identified based on the GSE117261 dataset. Weighted gene co-expression network analysis (WGCNA) was used to analyze the pre-processed data. Functional enrichment analysis was then carried out to explore the biological functions of these gene modules. The differentially co-expressed key gene modules were verified in-depth by GEO2R analysis. The immune infiltration in PAH was investigated by cell type identification by estimating relative subsets of RNA transcripts (CIBERSORT).
Results:
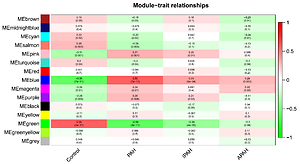
The WGCNA analysis found 15 differentially co-expressed gene modules, with the blue module indicating the strongest positive link to PAH and the green module indicating the strongest negative association with PAH. The kyoto encyclopedia of genes and genomes (KEGG) pathway analysis showed that the genes in the blue module were largely enriched in lysosome, complement, and coagulation cascades, and others, whereas the genes in the green module were primarily enriched in the chemokine signaling pathway, platelet activation, etc. Integrin subunit alpha M (ITGAM) was identified as the differentially co-expressed key gene. Immune infiltration analysis by CIBERSORT showed the differences between the PAH and control groups or between the PAH subgroups.
Conclusions:
ITGAM was considered a promising biomarker to discriminate PAH from the controls. Obvious differences were observed in immune infiltration between patients with the PAH and normal groups, providing new insight into understanding the molecular mechanisms on PAH’s pathogenesis.
Gene expression and immune cell infiltration are essential in the etiopathogenesis of pulmonary arterial hypertension (PAH). Our study attempted to identify novel differentially co-expressed genes and to investigate the features of immune cell infiltration in PAH.
Material and methods:
The GSE113439 and GSE117261 datasets were acquired from the Gene Expression Omnibus (GEO) database. The differentially expressed genes (DEGs) between the PAH and control groups were identified based on the GSE117261 dataset. Weighted gene co-expression network analysis (WGCNA) was used to analyze the pre-processed data. Functional enrichment analysis was then carried out to explore the biological functions of these gene modules. The differentially co-expressed key gene modules were verified in-depth by GEO2R analysis. The immune infiltration in PAH was investigated by cell type identification by estimating relative subsets of RNA transcripts (CIBERSORT).
Results:
The WGCNA analysis found 15 differentially co-expressed gene modules, with the blue module indicating the strongest positive link to PAH and the green module indicating the strongest negative association with PAH. The kyoto encyclopedia of genes and genomes (KEGG) pathway analysis showed that the genes in the blue module were largely enriched in lysosome, complement, and coagulation cascades, and others, whereas the genes in the green module were primarily enriched in the chemokine signaling pathway, platelet activation, etc. Integrin subunit alpha M (ITGAM) was identified as the differentially co-expressed key gene. Immune infiltration analysis by CIBERSORT showed the differences between the PAH and control groups or between the PAH subgroups.
Conclusions:
ITGAM was considered a promising biomarker to discriminate PAH from the controls. Obvious differences were observed in immune infiltration between patients with the PAH and normal groups, providing new insight into understanding the molecular mechanisms on PAH’s pathogenesis.
Share
RELATED ARTICLE
| eISSN: | 1896-9151 |
| ISSN: | 1734-1922 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.