Introduction
Pulmonary hypertension (PH) is a progressive disease of multifactorial etiology with poor prognosis due to right heart failure. It is defined by a resting mean pulmonary artery pressure (PAP) of 25 mm Hg or more. According to pathophysiological appearance the following subgroups of PH have been specified: primary pulmonary hypertension, called idiopathic or idiopathic pulmonary arterial hypertension (IPAH), and secondary pulmonary hypertension. The latter can be further divided into: passive – in the course of left, congestive heart failure, active – due to diseases characterized by hypoxia, poisoning, or in the case of vasoactive drug use, hyperkinetic – due to congenital heart defects with leakage from left to right heart, obstructive – in the course of collagenases and other disorders causing pressure on pulmonary capillaries, and in pulmonary embolism. Anatomopathologically, PH is characterized to be precapillary – arterial, post-capillary – venous or mixed.
The introduction of disease targeted therapies has significantly improved management and patient survival in the above-mentioned cases. Current guidelines propose use of two or more classes of drugs that may be applied sequentially or initially (upfront). Several randomized clinical trials and meta-analyses have revealed the efficacy of such PH-specific therapies toward reduction of clinical worsening, defined as a combination of death, admission to hospital, lung transplantation, symptomatic progression or treatment escalation including initiation of prostacyclins [1–6].
Adverse drug reactions (ADRs) with pulmonary vasodilator use in PH patients are usually reported in the Summaries of Product Characteristics (SmPCs) as common (i.e. 1/10–1/100 patients) or very common (i.e. ≥ 1/10 patients). They may contribute to a worse quality of life, prevent therapeutic escalation or precipitate discontinuation. In such cases, drug avoidance may in turn contribute to a worse prognosis. On the other hand, the presence of dose-related ADRs may suggest a higher circulating dose and, in some cases, be associated with improved mortality despite the impact on health-related quality of life [7].
It remains unknown whether the increased efficacy of such combinations of two or more agents targeting PH is accompanied by diminished safety. While there is a need to identify the efficacy-to-safety ratio of therapeutic strategies including combined regimens, there is no comprehensive analysis on the adverse event profiles of different targeted therapies in PH.
The objective of this study was to systematically review the safety of various disease-specific agents approved to manage PH, according to data reported in randomized, controlled clinical trials using both placebo-controlled and active comparators. We propose a comparative appraisal across particular medication classes, i.e.: endothelin receptor antagonist (ERA), prostacyclin analogues (PGI2), prostacyclin receptor (IP) agonists, phosphodiesterase type 5 inhibitors (PDE-5i) or guanylate cyclase stimulators (GCs). Besides the therapeutic group, other determinants, such as the amount of agents added to baseline therapy (i.e. 1, 2 or more) or route of administration, were taken into consideration.
Material and methods
Data source
Meta-analysis was reported in accordance with the PRISMA guidelines [8]. The search corpus comprised Medline, Scopus, Embase and Clinical Trials from January 1st 1990 to May 17th 2018. The databases were searched with no language restrictions using the following search terms in titles and abstracts: (‘bosentan’, OR ‘ambrisentan’, OR ‘epoprostenol’, OR ‘treprostinil’, OR ‘iloprost’, OR ‘selexipag’, OR ‘sildenafil’, OR ‘tadalafil’, OR ‘riociguat’, OR ‘macitentan’) AND ‘humans’, AND ‘pulmonary hypertension’, AND ‘clinical trials’. Two investigators evaluated each article independently. The selection of abstracts was independently carried out by two different researchers (K.S. and A.S.), and disagreements were resolved through discussion with a third researcher (M.J-S.). Next, the eligibility of selected papers/trials was confirmed after reading the complete text. Each article underwent independent, blinded, double-data extraction by two reviewers, discrepancies in data extraction underwent arbitration by a third reviewer and consensus was obtained by verbal discussion.
Study selection
Inclusion criteria were defined a priori. Original studies were included if they met the following criteria: (i) being a clinical prospective, double-blind, randomized trial assessing the effects of additional PH-targeted therapy with any dose compared with background (placebo, non-specific or specific) therapy in adult patients with PH, (ii) recruiting patients with a clinical diagnosis of PH (idiopathic – IPAH, connective tissue disease – CTD, due to human immunodeficiency virus – HIV infection or drug-induced pulmonary arterial hypertension – DPAH) as well as patients developing the venous form of pulmonary hypertension, e.g. due to congenital heart disease (CHD). In the placebo arm, only supportive therapy was considered: oral anticoagulants, diuretics, oxygen or digoxin and other cardiovascular drugs. The specific PH agents had to be withdrawn at least 3 months before enrolment.
Studies were excluded if any of the following criteria were met: 1. Lack of original data; 2. Lack of blinding; 3. Lack of control group (comparator); 4. No adverse reactions were reported/described by authors; 5. Animal studies.
Quality assessment
The information of methodological quality was extracted as well, in particular data on random sequence generation, blinding or indications of incomplete outcome information according to Jadad criteria (0–5 pts) [9]. The quality was assessed independently by two investigators. In case of any discrepancies a discussion was carried out to achieve a consensus.
The outcomes involved: (i) severe adverse drug reactions (SADRs), (ii) discontinuations because of ADR and (iii) particular adverse reactions that were classified according to the Medical Dictionary for Regulatory Activities (MedRA) (version 16). A structured form developed in MS Excel was used to extract data on trial and patient population characteristics and outcomes.
Statistical analysis
The meta-analysis was conducted using STATISTICA 13.1 software and Module for Meta-analysis (StatSoft, Poland). The difference of dichotomous data between two groups was estimated with relative risk (RR) with 95% two-tailed confidence intervals (CI). A DerSimonian and Laird random-effects model was used to compensate for the heterogeneity of studies [10]. We analyzed whether the risk of particular ADR increased when a PH-specific agent belonging to the following groups was added to the baseline (placebo or other PH targeted agent): ERA, prostacyclin and their analogues (PGI2), IP agonists, PDE-5i and soluble GCs. These findings were reported in terms of “RR parameter”. In order to examine the robustness of the results, sensitivity analysis was conducted using the leave-one-out method, i.e. removing one study each time and recalculating the results.
Subgroup analyses
Heterogeneity was quantitatively assessed using Cochran’s Q, and I² statistics. P < 0.05 was regarded as statistically significant. To explore the potential sources of heterogeneity, we performed particular subgroup analyses to determine the impact of route of administration (p.o., i.v., s.c., inh) and amount of agents added to baseline therapy (i.e. 1, 2 and more; monotherapy vs. combination) on the obtained outcome. Following this, all statistically significant ADRs were taken together and their risk calculated separately for monotherapy and combined regimens to determine whether combining two or more agents can significantly alter relative risk. The differences among compared factors were confirmed with a statistically significant measure of Qinter-group value (p < 0.05).
Potential publication bias was examined using a visual inspection of Begg’s funnel plot asymmetry, Begg’s rank correlation, and Egger’s weighted regression [11, 12]. Duval and Tweedie ‘trim and fill’ was used to adjust the analysis for the effects of publication bias [13].
Results
Characteristics of included trials
In total, 1701 articles were screened; 449 duplicates were removed, and another 837 were excluded because they did not meet inclusion criteria. Out of 415 eligible papers, 380 were excluded since they were not blinded (n = 168); they did not have comparator (n = 91); or the adverse events were not mentioned by the authors (n = 80). Other reasons included e.g. lack of original data, only non-specific or non-pharmacological therapy that was introduced for pulmonary hypertension. Finally, we included 35 articles in this meta-analysis (Figure 1). In one study (acronym: AMBITION) two arms were included separately, and 36 trials were analyzed, finally. The median Jadad score was three out of five (interquartile range [IQR]: 3–4) (Table I).

Table I
Characteristics of included studies
| Trial name | No. of patients | PH type (proportion %) | WHO-FC (%) | LengthR [weeks] | Baseline therapy (%) | Therapeutic arm | No. of drugs added to baseline | Outcomes/Jadad score | Rate of experiencing severe drug reaction | Rate of discontinuation# |
|---|---|---|---|---|---|---|---|---|---|---|
| AIR [23] | 203 | IPAH (50.25%) PAH-CTD (17.24%) DPAH (4.43%) CTEPH (28.08%) | III (58.62%) IV (41.38%) | 12 | Placebo (100%) | Iloprost 15–45 µg (inh) | 1 | Primary: increase of at least 10% in the 6MWD; WHO-FC in the absence of a deterioration in the clinical condition or death; Secondary: 6MWD; WHO-FC; dyspnea rating; hemodynamic measurements; QoL; clinical deterioration; death; and the need for transplantation/3 | SADRs 53/203; other ADRs 181/203 | 18/203 |
| AMBITION [24]** | 500 | IPAH (53.0%) PAH-CTD (37.4%) PH-CHD (1.8%) PAH-HIV (1.8%) DPAH (3.2%) | II (31%) III (69%) | 78 | Ambrisentan 10 mg Tadalafil 40 mg | Ambrisentan 10 mg plus Tadalafil 40 mg | 2 | Primary: time-to-event analysis of first event of clinical failure; Secondary: change from baseline to 24 wk. in: NT-proBNP level; 6MWD; WHO-FC; BDS/4 | SADRs 187/500 | 181/500 |
| ARIES-1 [25] | 201 | IPAH (63.00%) PAH-CTD (31.00%) PAH-HIV (3.50%) DPAH (2.50%) | I (2.49%) II (32.34%) III (58.21%) IV (6.96%) | 12 | Placebo (100%) | Ambrisentan 5–10 mg | 1 | Primary: 6MWD; Secondary: clinical worsening; WHO-FC; QoL; BDS; NT-proBNP/4 | SADRs 47/393 | 18/201 |
| ARIES-2 [25] | 192 | IPAH (65.10%) PAH-CTD (32.29%) PAH-HIV (2.08%) DPAH (0.53%) | I (1.56%) II (44.80%) III (51.56%) IV (2.08%) | 12 | Placebo (100%) | Ambrisentan 2.5–5 mg | 1 | Primary: 6MWD; Secondary: clinical worsening; WHO-FC; QoL; BDS; NT-proBNP/4 | 22/192 | |
| ARTEMIS-PH [26] | 40 | PAH and idiopathic cystic fibrosis (100%) | No data | 56 | Placebo (100%) | Ambrisentan 5–10 mg (p.o.) | 1 | Primary: 6MWD; Secondary: BDS; WHO-FC, pulmonary function tests, QoL; NT-proBNP/3 | SADRs 15/40; other ADRs 30/40N | 36/40N |
| Badesch et al. 2002 [27] | 32 | IPAH (84.38%) PAH-CTD (15. 62%) | III (100%) | 12 | Placebo (100%) | Bosentan 62.5 mg BID (for 4 weeks) 125 mg BID | 1 | Primary: change in exercise capacity; Secondary: cardiopulmonary hemodynamics; BDS; WHO-FC; withdrawal due to clinical worsening/3 | ADRs (total) 20/32 | 3/32 |
| BREATHE-1 [28] | 213 | IPAH (70.42%) PAH-CTD (29.58%) | III (91.55% IV (8.45%) | 16 | Placebo (100%) | Bosentan 62.5 mg BID (for 4 weeks) 125–250 mg BID | 1 | Primary: degree of change in exercise capacity; Secondary: BDS; WHO-FC; TTCW/3 | 174/213* | 14/213 |
| BREATHE-2 [29] | 33 | IPAH (81.8%) PAH-CTD (18.2%) | III (75.8%) IV (24.2%) | 16 | Epoprostenol 2–16 ng/kg/min (i.v.) (100%) | Bosentan 62.5, 125 mg BID | 2 | Primary: TPR; Secondary: CI; PVR; mPAP; mRAP; 6MWD; dyspnea-fatigue rating; WHO-FC/4 | SADRs 5/33 | 5/33 |
| BREATHE-5 [30] | 54 | PH-CHD (100%) | III (100%) | 16 | Placebo (100%) | Bosentan 62.5–125 mg | 1 | Primary: systemic pulse oximetry; PVR; Secondary: 6MWD; hemodynamic measurements; functional capacity and safety/4 | ADRs 9 SADRs 8/54 | 4/54 |
| COMPASS-2 [31] | 334 | IPAH (63.8%) PAH-CTD (26.3%) PH-CHD (6.0%) DPAH (2.4%) | II (41.9%) III (57.5%) IV (0.6%) | 16 | Sildenafil ≥ 20 mg TID (100%) | Bosentan 125 mg BID | 1 | Primary: time to first morbidity/mortality; Secondary: 6MWD; WHO-FC; time to first occurrence of death from any cause; hospitalization for PAH; start of intravenous prostanoid therapy; atrial septostomy; lung transplant; death from any cause; NT-proBNP/3 | SADRs 175/334 other ADRs 282/334N | 172/334N |
| Denton et al. 2006 [32] | 66 | PAH-CTD (100%) | III (95.45%) IV (4.54%) | 16 | Placebo (100%) | Bosentan 125–500 mg | 1 | Primary: 6MWD; Secondary: WHO-FC; TTCW/3 | ADRs (incl. SADRs) 26 | 23/66 |
| DILATE [33] | 39 | PH-CHD (100%) | No data | 16 | Placebo (100%) | Riociguat 0.5–2 mg | 1 | Primary: the peak decrease from baseline mPAP; Secondary: hemodynamic and echocardiographic measurements, biomarker levels, safety variables, and pharmacokinetics/3 | SADRs 5/39 ADRs (total) 15/39 | 2/39 |
| EARLY [34] | 185 | IPAH (60.5%) PH-CHD (17.3%) PAH-CTD (17.8%) PAH-HIV (3.8%) Other (0.5%) | II (100%) | 24 | Treatment naïve (84%) Sildenafil (16%) | Bosentan 62.5–125 mg BID | 1 | Primary: PVR at rest; 6MWD; Secondary: TTCW; WHO-FC; BDS; hemodynamic measurements/4 | ADRs 111 SADRs 20/185 | 22/185 |
| FREEDOM-M [35] | 349 | IPAH (74.43%) PAH-CTD (19.25%) PH-CHD (5.17%) PAH-HIV (1.15%) | I (3.44%) II (35.82%) III (60.74%) | 12 | Placebo (100%) | Treprostinil 0.25–1 mg (p.o.) | 1 | Primary: 6MWD; Secondary: BDS; combined 6MWD/BDS; dyspnea-fatigue index; WHO-FC; symptoms of PAH; clinical worsening; and safety/3 | SADRs 67/349 ADRs (total) 325/349 | 69/349 |
| FREEDOM-C [36] | 350 | IPAH (66.3%) PAH-CTD (26.3%) PH-CHD (6.3%) PAH-HIV (1.1%) | I (0.9%) II (20.6%) III (76%) IV (2.6%) | 16 | ERA (30.3%) PDE-5i (25.1%) ERA and PDE-5i (44.6%) | Treprostinil 0.5–16 mg BID (p.o.) | 1 | Primary: 6MWD; Secondary: TTCW; combined ranking of 6MWD and BDS; dyspnea-fatigue rating/3 | SADRs 65/350 other ADRs 330/350N | 63/350 |
| FREEDOM-C2 [37] | 310 | IPAH (65.5%) PAH-CTD (31.3%) PAH-HIV (1.9%) PH-CHD (1.3%) | II (25.8%) III (72.6%) IV (1%) | 16 | ERA (17.1%) PDE-5i (42.6%) ERA and PDE-5i (40.3%) | Treprostinil 0.25–16 mg BID (p.o.) | 1 | Primary: 6MWD; Secondary: clinical worsening; BDS; combined 6MWD and BDS; NT-proBNP; WHO-FC/3 | SADRs 46/310 other ADRs 239/310N | 40/310 |
| GRIPHON [38] | 1156 | IPAH (55.4%) PAH-CTD (28.9%) PH-CHD (9.5%) PAH-HIV (0.9%) DPAH (2.3%) | I (0.8%) II (45.8%) III (52.5%) IV (1.0%) | 71 | Treatment naïve (20.4%) ERA (14.7%) PDE-5i (32.4%) ERA and PDE-5i (32.5%) | Selexipag (200–1600 µg) BID | 1 | Primary: time to event analysis of death or complication related to PAH; Secondary: 6MWD; absence of worsening of WHO-FC; death due to PAH or hospitalization for worsening of PAH; death from any cause; NT-proBNP/4 | SADRs 524/1156 other ADRs 1030/1156N | 615/1156N |
| Iversen et al. 2009 [39] | 21 | PH-CHD (Eisenmenger syndrome) (100%) | II (43%) III (48%) IV (5%) | 24 | Bosentan 62.5; 125 mg BID (100%) | Sildenafil 25–50 mg BID | 1 | Primary: 6MWD; Secondary: hemodynamic measurements; WHO-FC; NT-proBNP/3 | ADRs (total) 3/21 | 1/21 |
| Machado et al. 2011 [40] | 74 | Other (SCD) (100%) | No data | 16 | Placebo (100%) | Sildenafil 60–240 mg | 1 | Primary: 6MWD; Secondary: TRV; hemodynamic measurements; BDS; WHO-FC; NT-proBNP; QoL/3 | SADRs 25/74 other ADRs 58/74N | 45/74 |
| PACES [41] | 265 | IPAH (79.4%) PAH-CTD (11.6%) Other (3.7%) | I (1.1%) II (25.5%) III (65.5%) IV (6.0%) | 16 | Epoprostenol 3–181 ng/kg/min (i.v.) | Sildenafil 20–80 mg TID | 1 | Primary: 6MWD; Secondary: hemodynamic measurements; TTCW; BDS after completing the 6MWD/4 | SADRs 68/265 ADRs (total) 252/265 | 21/265 |
| PATENT PLUS [42] | 18 | IPAH (50%) PAH-CTD (33.3%) PH-CHD (11.1%) Other (5.6%) | I (5.6%) II (55.6%) III (33.3%) IV (5.6%) | 12 | Sildenafil 20 mg TID (100%) | Riociguat 1–2.5 mg TID | 1 | Primary: maximum change in supine SBP; Secondary: maximum change in standing SBP; supine and standing HR; DBP; changes in: 6MWD; WHO-FC; NT-proBNP; BDS; TTCW/4 | SADRs 2/18 other ADRs 16/18 | 1/18 |
| PATENT-1 [43] | 443 | IPAH (61.4%) PAH-CTD (25%) PH-CHD (8.0%) DPAH (1.0%) Other (3.0%) | I (3.0%) II (42%) III (53%) IV (1.0%) | 12 | Treatment naïve (50%) ERA (44%) Non-parenteral Prostanoids (6%) | Riociguat 1.5 or 2.5 mg TID | 1 | Primary: 6MWD; Secondary: pulmonary vascular resistance; NT-proBNP; WHO-FC; TTCW; BDS; QoL/4 | SADRs 63/443 other ADRs 369/443N | 40/443N |
| Pfizer study No. A1481243 2013 [44] | 103 | PAH (% – no data) | II (34.0%) III (65.0%) IV (1.0%) | 12 | Bosentan 62.5 or 125 mg BID | Sildenafil 20 mg TID | 1 | Primary: 6MWD; Secondary: WHO-FC; clinical worsening events; BDS; 1 year survival probability from the start of sildenafil treatment; 1 year survival from the start of sildenafil treatment/3 | SADRs 45/103 other ADRs 71/103N | 12/103N |
| PHIRST [45] | 405 | IPAH (61%) DPAH (3.9%) PAH-CTD (23.5%) Other (11.6%) | I (1.0%) II (32.1%) III (65.2%) IV (1.7%) | 16 | Bosentan (53.3%) Treatment naïve (46.7%) | Tadalafil 2.5, 10, 20, 40 mg | 1 | Primary: 6MWD; Secondary: WHO-FC; TTCW; BDS; QoL; hemodynamic measurements/3 | ADRs (total) 353/405 | 64/405 |
| Rubenfire et al. 2007 [46] | 22 | IPAH (72.73%) PAH-CTD (13.64%) PH-CHD (4.55%) | I (4.55%) II (54.55%) III (40.90%) | 8 | Placebo (100%) | Treprostinil 22–32 ng/kg/min (s.c.) | 1 | Primary: TTCW; Secondary: 6MWD; BDS; dyspnea-fatigue rating; symptoms of PAH; and cardiovascular hospitalizations/3 | ADRs (total) 22/22 | 9/22 |
| Sastry et al. 2004 [47] | 22 | IPAH (100%) | II (81.82%) III (18. 18%) | 12 | Placebo (100%) | Sildenafil 25–100 mg TID | 1 | Primary: change in exercise time; Secondary: changes in pulmonary artery systolic pressure and cardiac output, QoL/4 | ADRs 61 SADRs – none | 2/22 |
| SERAPHIN [48] | 742 | IPAH (54.4%) PAH-CTD (30.5%) PH-CHD (8.4%) PAH-HIV (1.4%) DPAH (3.0%) | I (0.1%) II (52.%) III (45.6%) IV (1.9%) | 104 | Treatment naïve (33.2%) PDE-5i (61.4%) Prostanoids (5.4%) | Macitentan 3 mg, 10 mg | 1 | Primary: time to first event related to PAH or death from any cause; Secondary: 6MWD; WHO-FC; hospitalization for PAH; death from any cause; safety/5 | SADRs 376/742 other ADRs 668/742N | 155/742N |
| Simonneau et al. 2002 [49] | 469 | IPAH (57.57%) PH-CHD (23.24%) PAH-CTD (19.19%) | II (11.30%) III (81.45%) IV (7.25%) | 12 | Placebo (100%) | Treprostinil 1.25–22.5 ng/kg/min (s.c.) | 1 | Primary: 6MWD; Secondary: BDS; cardio-pulmonary hemodynamics measurements; QoL/4 | ADRs 11 SADRs – none | 29/469 |
| Simonneau et al. 2012 [50] | 43 | IPAH (76.8%) DPAH (4.7%) PAH-CTD (14.0%) PH-CHD (4.7%) | II (39.5%) III (60.5%) | 17 | ERA (37.2%) Sildenafil (27.9%) ERA and Sildenafil (34.9%) | Selexipag 200–800 µg BID | 1 | Primary: PVR; Secondary: hemodynamic measurements; 6MWD; TTCW; BDS; WHO-FC; NT-proBNP/4 | SADRs 10/43 ADRs (total) 41/43 | 4/43 |
| STEP [51] | 67 | IPAH (55%) APAH (45%) | II (1.5%) III (94%) IV (4.5%) | 12 | Bosentan 125 mg BID (100%) | Iloprost 2.5–5 µg 6–9 times daily (inh) | 1 | Primary: 6MWD; Secondary: WHO-FC; BDS; hemodynamic measurements; TTCW/5 | SADRs 12/67 any of ADR 64/67 | 9/67 |
| Wirostko et al. 2012 [52] | 277 | IPAH (63.18%) PAH-CTD (30.32%) PH-CHD (6.50%) | I (0.36%) II (38.63%) III (57.76%) IV (3.25%) | 12 | Placebo (100%) | Sildenafil 60–240 mg | 1 | Ocular safety/4 | ADRs 100/277 SADRs – none | 12 |
| SUPER-1 [53] | 277 | IPAH (63.18%) PAH-CTD (30.32%) PH-CHD (6.50%) | I (0.36%) II (38.63%) III (57.76%) IV (3.25%) | 12 | Placebo (100%) | Sildenafil 20–80 mg TID | 1 | Primary: 6MWD; Secondary: mPAP; BDS; WHO-FC; TTCW/4 | ADRs 371 SADRs 2/277 | 12/277 |
| TRIUMPH [54] | 235 | IPAH (55.7%) PAH-CTD (32.8%) Other (11.5%) | III (97.9%) IV (2.1%) | 12 | Bosentan 125 mg (70.2%) Sildenafil ≥ 20 mg TID (29.8%) | Treprostinil 18–54 µg QID (inh) | 1 | Primary: peak 6MWD; Secondary: TTCW; BDS; WHO-FC through 6MWD at 12 weeks; peak 6MWD at 6 weeks; QoL; NT-proBNP/3 | SADRs 22/235 other ADRs 201/235N | 23/235N |
| TRUST-1 [55] | 44 | IPAH (95.00%) PAH-CTD (5.00%) | III (95.00%) IV (5.00%) | 12 | Placebo (100%) | Treprostinil 4–14 ng/kg/min (i.v.) | 1 | Primary: 6MWD; Secondary: BDS; Dyspnea-Fatigue Index; WHO-FC; Clinical Worsening; combined 6MWD/BDS/3 | SADRs 19/44 ADRs (total) 43/44 | 13/44 |
| VISION [56] | 67 | PAH (% – no data) | No data | 16 | Sildenafil Bosentan | Iloprost 5 µg 4–6 times daily (inh) | 1 | Primary: 6MWD; Secondary: change in 6MWD measured after inhalation following 16 weeks of combination therapy/3 | SADRs 11/67 other ADRs 60/67 | 9/67 |
| Zhuang et al. 2014 [57] | 124 | IPAH (62.9%) DPAH (8.9%) PAH-CTD (22.6%) PH-CHD (5.6%) | I (< 1%) II (57.3%) III (38.7%) IV (4.0%) | 16 | Ambrisentan 10 mg (100%) | Tadalafil 40 mg | 1 | Primary: 6MWD; Secondary: WHO-FC; TTCW; hemodynamic measurements/3 | ADRs 148 SADRs – no data | 11/124 |
6MWD – 6-minute walk distance, BID – two times a day, BDS – Borg dyspnea score, CI – cardiac index, DPAH – drug-induced pulmonary arterial hypertension, DPB – diastolic blood pressure, ERA – endothelin receptor antagonists, HPAH – heritable pulmonary arterial hypertension, HR – heart rate, IPAH – idiopathic pulmonary arterial hypertension, mPAP – mean pulmonary artery pressure, mRAP – mean right atrial pressure, NT-proBNP – N-terminal pro-brain natriuretic peptide, PAH-CTD – PAH associated with connective tissue disease, PAH-HIV – PAH associated with HIV, PH-CHD – PH associated with congenital heart disease, SCD – sickle cell disease, PDE-5i – phosphodiesterase type-5 inhibitors, PVR – pulmonary vascular resistance, QID – four times a day, QoL – quality of life, SADR – severe adverse drug reaction, SBP – systolic blood pressure, TID – three times a day, TPR – total pulmonary resistance, TTCW – time to clinical worsening, WHO-FC – World Health Organization functional class,
# due to any cause (e.g. adverse event, death, study termination by the investigator/sponsor, lost to follow-up, lung transplantation and others), Ndata from clinicaltrials.gov (NCT), total – ADRs + SADRs, Rrandomized study without open-label extension was considered, rate of experiencing SADRs/ADRs was for both investigational group and placebo.
All the analyzed studies were randomized, double-blinded and placebo controlled. This meta-analysis comprised 7977 PH patients: 4674 with iPAH, 2082 with CTD-PAH, 595 with pulmonary hypertension due to CHD, 112 with DPAH, and 514 with other forms of PH, including idiopathic pulmonary fibrosis, sickle cell disease, portopulmonary hypertension or PH associated with atrial septal defect.
Among them we assessed the increased risk of adverse effects resulting from addition to the background (placebo or aimed therapy) of the following targeted strategies: (i) endothelin receptor antagonist (ERA): bosentan, ambrisentan or macitentan (n = 3590 patients); (ii) phosphodiesterase type 5 inhibitor (PDE-5i): sildenafil or tadalafil (n = 3675); (iii) soluble guanylate cyclase stimulator (GCs): riociguat (n = 354); (iiii) prostacyclins (PGI2): treprostinil (p.o., i.v., s.c.), or iloprost (inh) (n = 1515) or selective prostacyclin receptor agonist (selexipag) (n = 607) within a 2 to 26 month-period. The primary and secondary outcomes included: 6-minute walk distance (6MWD); time to clinical worsening (TTCW); hemodynamic parameters: systolic (diastolic) blood pressure; heart rate (HR) and pulmonary vascular resistance (PVR) or N-terminal pro-brain natriuretic peptide (NT-proBNP) (Table I).
Discontinuations and severe drug reactions
Discontinuations because of adverse events were reported in 30 per 36 trials included in the meta-analysis. Overall, no significant impact on the increased risk of discontinuations due to ADR was identified (RR = 0.99, 95% CI: 0.74–1.32, p = 0.9443, I2 = 50.88%). Further subgroup analysis revealed some increased risk only when the IP agonist selexipag was added to the baseline therapy (RR = 2.01, 95% CI: 1.41–2.86, p < 0.0001, Qinter-group, 17.53, df = 4, p = 0.0015), not the other agents (Figure 2 A). Route of administration or therapeutic regimen was also not found to have any significant impact on the altered risk. Patients receiving the combined regimen demonstrated a greater tendency of risk cessation (Qinter-group, p = 0.0779). Such risk of discontinuation was increased particularly when bosentan (ERA) (RR = 1.64, 95% CI: 1.11–2.43, p < 0.05), treprostinil (PGI2) (RR = 3.27, 95% CI: 1.28–8.34, p < 0.05) or selexipag (IP agonist) (RR = 2.01, 95% CI: 1.41–2.86, p < 0.05) was added to the baseline (Qinter-group, p < 0.05) (Figure 2 B).
Figure 2
Effect of pulmonary hypertension (PH)-specific agents on the relative risk (RR, 95% CI): A – Discontinuations due to adverse drug reactions (ADRs) – RR = 0.99, 95% CI: 0.74–1.32, p = 0.9443, I2 = 50.88%; B – Discontinuations for combined regimens (subgroup analysis). The relative risk of discontinuation was increased particularly when bosentan, treprostinil or selexipag was added to the monotherapy; C – Severe ADRs – RR = 0.97, 95% CI: 0.84–1.13, p = 0.7210; D – Categories of ADRs* (bold font) with selected subcategories. For combination regimens, a significant tendency toward increased RR was observed for such ADRs as blood and lymphatic system disorders with anemia subgroup, gastrointestinal disorders with diarrhea and nausea subgroups, myalgia and pain in limb, respiratory, vascular or nervous system disorders with headache subgroup. M – monotherapy (comparator: placebo/supportive therapy), C – combined therapy (active comparator: monotherapy). *only ADRs with significant result for relative risk were examined
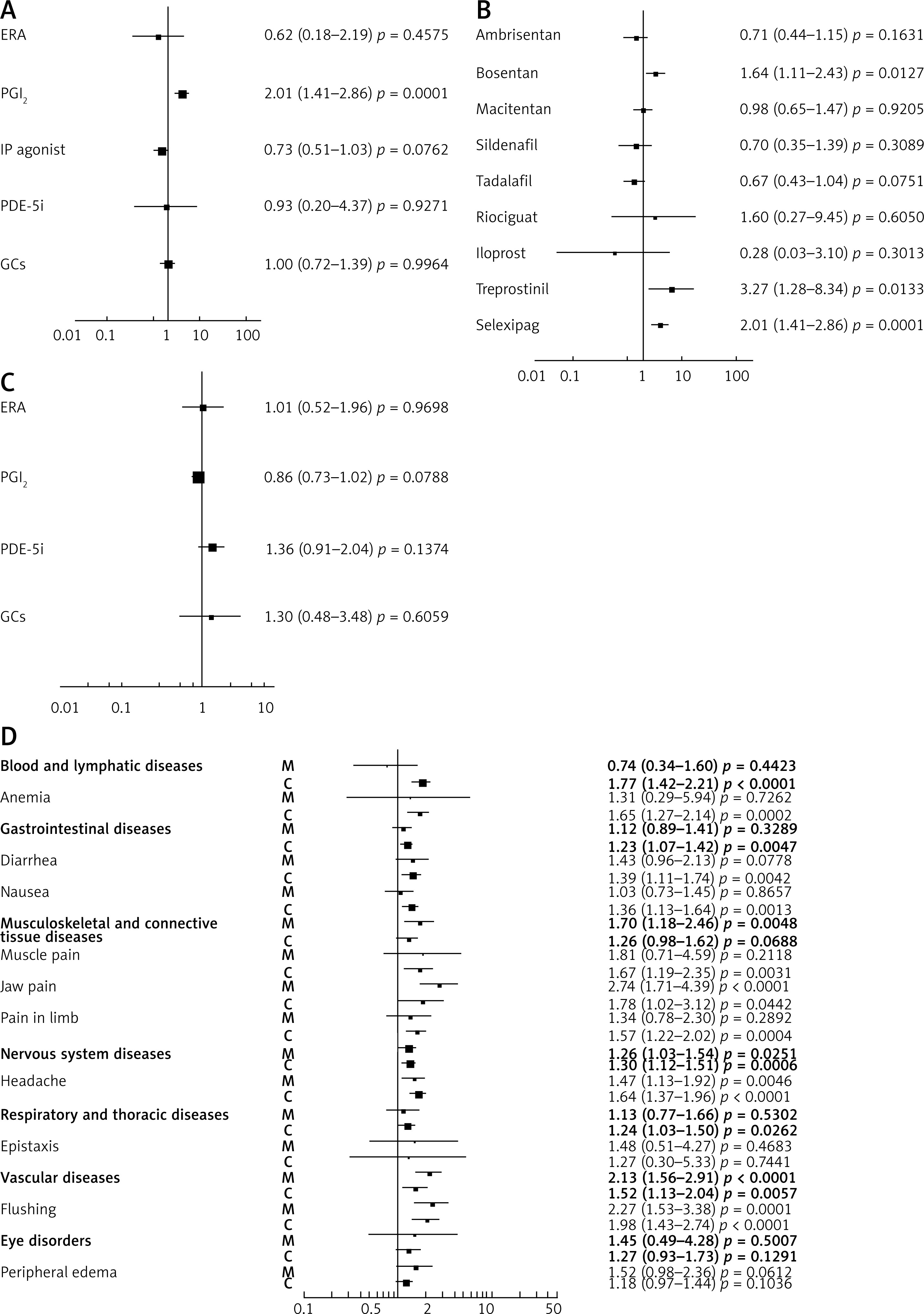
Severe drug reactions were reported in 25 out of 36 trials. No significant impact on the increased risk of severe ADR was found (RR = 0.97, 95% CI: 0.84–1.13, p = 0.7210, I2 = 25.84%). We did not observe any discrepancies according to particular agent or therapeutic group (Figure 2 C). No other factors such as route of administration or therapeutic regimen (i.e. one, two or three drugs added to baseline) were revealed to determine the effect size.
Adverse drug reactions (other than severe ADRs)
When considering all significant ADRs as a whole, subjects who used monotherapy experienced increased risk of adverse reactions (RR = 1.44, 95% CI: 1.27–1.64, p < 0.0001; comparator: placebo/supportive therapy). Similarly, in patients receiving combined therapy the relative risk was significantly increased (RR = 1.42, 95% CI: 1.32–1.54, p < 0.0001; active comparator – PH-specific agent). A significant tendency toward increased RR was observed for such ADRs as blood and lymphatic system disorders with the anemia subgroup, gastrointestinal disorders with diarrhea and nausea subgroups, respiratory and thoracic diseases, nervous system disorders with headache, vascular events, myalgia and pain in limb. The first four main categories of ADRs tended to occur more often in combination regimens as compared to monotherapy (Figure 2 D).
The overall frequency for events classified into the remaining categories (e.g. blood and lymphatic system, the gastrointestinal, musculoskeletal or nervous systems, or respiratory or vascular disorders) was confirmed to be high (SmPC categories: common or very common). Figure 3 presents results for ADRs. A detailed description is provided in Supplementary Material (Results).
Figure 3
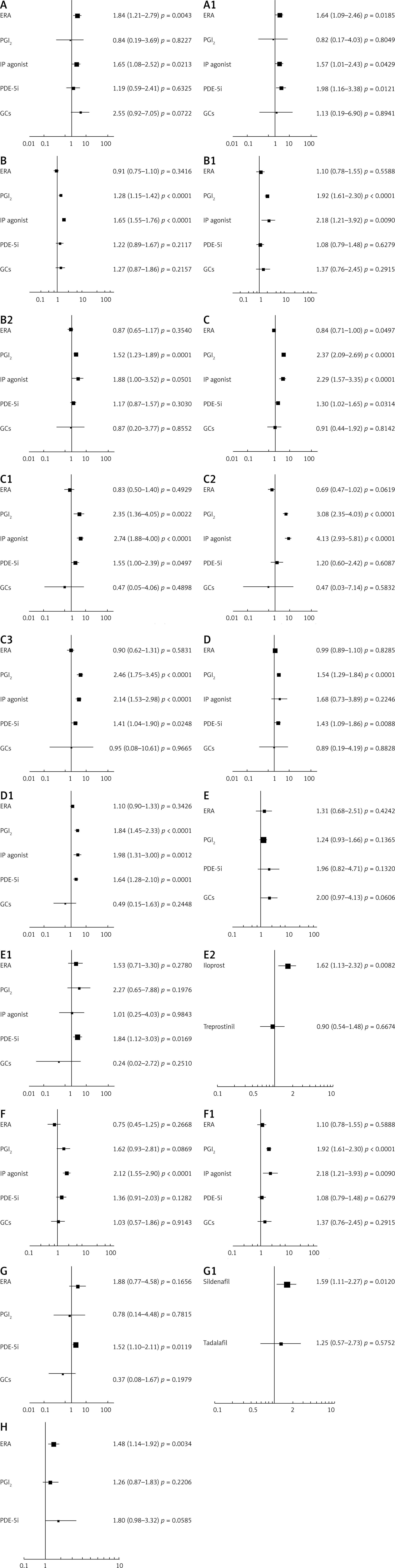
Effect of pulmonary hypertension (PH)-specific agents on the relative risk (RR, 95% CI) of selected adverse drug reactions (ADRs). The overall frequency of ADRs in the investigational groups was as follows: A – blood and lymphatic disorders (monotherapy: 8.53% (baseline)/4.22% (comparator) and combination: 4.74% (baseline)/9.74% (comparator), A1 – anemia (1.55%/2.20% and 3.38%/6.19%), B – gastrointestinal disorders (25.80%/32.19% and 55.40%/69.57%), B1 – diarrhea (11.20%/16.59% and 15.51%/20.67%), B2 – nausea (14.51%/17.30% and 14.99%/18.49%), C – musculoskeletal and connective tissue disorders (11.44%/26.89% and 34.61%/43.54%), C1 – muscle pain (1.69%/5.07% and 4.12%/7.30%), C2 – jaw pain (3.69%/12.18% and 4.40%/10.47%). C3 – pain in limb (7.47%/10.67% and 7.31%/11.09%), D – nervous system disorders (27.70%/37.39% and 46.83%/58.79%), D1 – headache (20.37%/32.04% and 22.34%/32.95%), E – respiratory and thoracic system disorders (19.82%/19.73% and 56.26%/70.40%), E1 – epistaxis 1.27%/4.02% and 2.58%/3.62%), E2 – cough (sub-group analysis: PGI2), F – vascular disorders (5.79%/11.22% and 9.90%/17.14%), F1 – flushing (4.75%/10.75% and 6.02%/12.09%). G – eye disorders (8.74%/14.91% and 0.84%/2.05%), G1 – subgroup analysis: PDE-5i, H – peripheral edema (7.26%/12.58% and 12.65%/15.49%). Significantly increased risk was denoted for overall category (A) and for the anemia events subcategory (A1). The risk of any gastrointestinal disorders was slightly increased (B) with predominant contribution of prostacyclins (PGI2) or IP agonist. The most prevalent were diarrhea (B1) and nausea (B2). The risk of musculoskeletal system-linked events was increased in a slight but statistically significant manner (C). Muscle pain, jaw pain and pain in limb were reported to significantly increase for selexipag and PGI2s (C1–C3). In accordance with nervous system (D) or headache subcategory (D1) increased RR was observed when PGI2 or PDE-5i was added to baseline. Overall, the relative risk of respiratory and thoracic disorders was slightly, but significantly increased (E). Incidents of epistaxis (E1) mainly involving sildenafil were noted. Some increase of cough incidence in the subgroup of patients receiving iloprost was noted (E2). PGI2s and selexipag were particularly superior to other agents for enhancing risk of vascular disorders (overall) (F) and flushing (F1). The risk of events classified as ‘eye disorders’ was slightly but significantly increased with the predominant contribution of PDE-5i: sildenafil (G–G1). An increased risk of peripheral edema was noted when ERAs were added to the baseline (H)
Publication bias
A visual inspection of funnel plots and Egger’s test were used to evaluate publication bias (Figure 4).
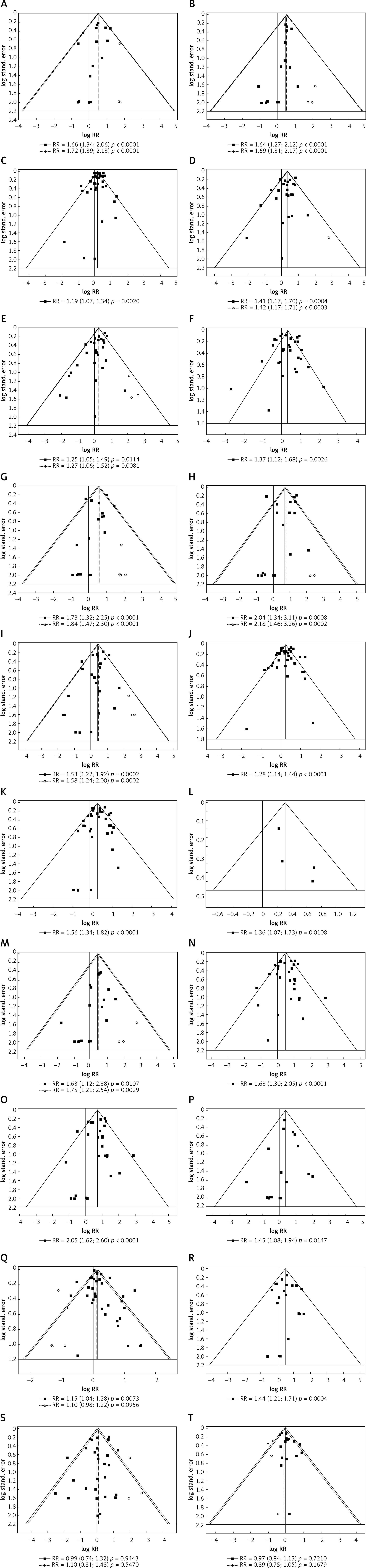
Figure 4
Publication bias of meta-analysis by Trim and Fill analysis. Funnel plots (RR – relative risk, random effect – 95% CI) show the distribution of published study outcomes (filled squares) vs. unpublished outcomes (open circles) estimated by Trim and Fill analysis. Dashed line represents RR and 95% CI with the added potentially unpublished studies and solid line represents published studies included in meta-analysis. Vertical dashed line represents the global estimate of safety. A – Blood and lymphatic disorders, B – anemia, C – gastrointestinal disorders, D – diarrhea, E – nausea, F – musculoskeletal and connective tissue disorders. G – muscle pain, H – jaw pain, I – pain in limb, J – nervous system disorders, K – headache, L – respiratory and thoracic system disorders. M – epistaxis, N – vascular disorders, O – flushing, P – eye disorders, Q – general disorders and administration site conditions, R – peripheral edema, S – discontinuations and T – SADRs. Non-significant result of Egger’s test for A, C–G, I–P and R. According to visual inspection and results of Egger’s test there is a suggestion of missing studies and publication bias excluding C, F, J, K, L, N, O, P, R Result of Egger’s test for (S) and (T) was non-significant. However, in the middle and right of the plot (S), and in the top diagram (T) there is a suggestion of missing studies, making publication bias plausible
Sensitivity analysis
In relation to the majority of reported ADRs, the observed effect was robust in sensitivity analysis and the statistical significance was not influenced by any single study included in the meta-analysis. Table II summarizes the sensitivity analysis with the most frequent subcategories of ADRs that were reported in studies included in the meta-analysis.
Table II
Summary of sensitivity analysis. The analysis was performed to exclude potential studies with the biggest RR outlier. In cases where any single study did not influence the statistical significance of the final outcome, all trials were included in the analysis. ID – MedRA codes: categories with selected subcategories
| Category | ID | Studies removed |
|---|---|---|
| Blood and lymphatic system disorders: | 10005329 | All studies included |
| Anemia | 10002034 | All studies included |
| Cardiac disorders: | 10007541 | All studies included |
| Palpitation | 10033556 | All studies included |
| Right ventricular failure | 10039163 | All studies included |
| Congenital, familial and genetic disorders | 10010331 | All studies included |
| Ear and labyrinth disorders | 10013993 | All studies included |
| Endocrine disorders | 10014698 | All studies included |
| Eye disorders | 10015919 | All studies included |
| Gastrointestinal disorders: | 10017947 | All studies included |
| Diarrhea | 10012735 | All studies included |
| Nausea | 10028813 | All studies included |
| Vomiting | 10047700 | All studies included |
| General disorders and administration site conditions: | 10018065 | All studies included |
| Fatigue | 10016256 | All studies included |
| Peripheral edema | 10034570 | 2 studies removed [38, 43] |
| Hepatobiliary disorders | 10019805 | All studies included |
| Immune system disorders | 10021428 | All studies included |
| Infections and infestations: | 10021881 | All studies included |
| Upper respiratory tract infections | 10046309 | 1 study removed [38] |
| Injury, poisoning and procedural complications | 10022117 | All studies included |
| Investigations | 10022891 | 1 study removed [31] |
| Metabolism and nutrition disorders | 10027433 | All studies included |
| Musculoskeletal and connective tissue disorders: | 10028395 | 1 study removed [31] |
| Arthralgia | 10003246 | All studies included |
| Jaw pain | 10023157 | All studies included |
| Muscle pain | 10028322 | All studies included |
| Pain in limb | 10033447 | All studies included |
| Neoplasms benign, malignant and unspecified (incl. cysts and polyps) | 10029104 | All studies included |
| Nervous system disorders: | 10029205 | All studies included |
| Headache | 10019211 | All studies included |
| Dizziness | 10013573 | All studies included |
| Syncope | 10042772 | All studies included |
| Pregnancy, puerperium and perinatal conditions | 10036585 | All studies included |
| Psychiatric disorders | 10037175 | All studies included |
| Renal and urinary disorders | 10038359 | All studies included |
| Reproductive system and breast disorders | 10038604 | All studies included |
| Respiratory, thoracic and mediastinal disorders: | 10038738 | 2 studies removed [56, 57] |
| Cough | 10011224 | 1 study removed [31] |
| Dyspnea | 10013968 | All studies included |
| Epistaxis | 10015090 | 1 study removed [36] |
| Skin and subcutaneous tissue disorders: | 10040785 | 3 studies removed [27, 38, 43] |
| Rash | 10037844 | 1 study removed [24] |
| Social circumstances | 10041244 | All studies included |
| Surgical and medical procedures | 10042613 | All studies included |
| Vascular disorders: | 10047065 | All studies included |
| Flushing | 10016825 | All studies included |
Discussion
At the time of writing, this was the first meta-analysis of 7977 participants intended to compare the safety profile of particular PH-specific therapies according to individual risk of adverse drug reaction based on a review of randomized trials using both placebo-controlled and active comparators.
Our main finding is that individual PH-targeted therapies do not significantly enhance the risk of either discontinuations due to ADRs or severe adverse drug reactions. The risk of cessation was comparable between patients receiving PH-specific agents in monotherapy or combination, with some tendency toward the combined regimen. In the subgroup of patients receiving combined therapy, the relative risk of discontinuation due to adverse events was significantly increased when treprostinil, selexipag or bosentan was added to the baseline. Likewise, in a previously published meta-analysis, it was found that treatment discontinuation was more likely to occur in patients receiving combined therapy, and this increased risk was particularly pronounced for non-parenteral prostaglandins and the selective prostacyclin receptor agonist [6]. As presented in Table II, the overall rate of discontinuations observed in some trials was high, about 30–50%. These figures included events from any cause, with the most common ones being termination by sponsor or investigator (ARTEMIS-PH), withdrawal of consent (COMPASS-2), morbidity or mortality primary endpoint (GRIPHON), as well as loss of follow-up or lung transplantation, while the percentage of discontinuations due to adverse events was lower (5–15%).
Both the present study and a previous one [14] suggest that the incidence of serious adverse events was similar between the monotherapy and combination regimen.
Of 26 main categories of ADRs defined by the MedRA Dictionary, 18 were not significantly affected by particular therapeutic agents as compared to baseline; these included benign neoplasms, disorders of the reproductive system and breast, infections or immune system disorders, or those linked to the endocrine system, kidney and urinary tract.
Conversely, the overall frequency for events classified into the remaining categories (e.g. blood and lymphatic system, the gastrointestinal, musculoskeletal or nervous systems, or the respiratory or vascular disorders) were confirmed to occur with high frequency (> 1/10 or > 1/100). Particular PH-specific therapies had the greatest effect on events associated with vasodilatory activity: flushing and headache as well as jaw pain, limb pain and myalgia. Musculoskeletal adverse events are common ones related to therapy with prostanoid (PGI2) or IP receptor agonists. Prostanoid-based therapy remains a critical component of optimal PH management, particularly for patients with the most severe disease. We can demonstrate a two- or three-fold increase in the risk of myalgia or jaw pain in cases where inhaled iloprost, treprostinil (both: p.o. and s.c.) or selexipag was added to the baseline. Current international guidelines recommend them both in non-vasoreactive and treatment-naive patients at high risk, with initial combination therapy including intravenous epoprostenol (class A, level I) or prostacyclin analogues. Selexipag is an orally administered IP receptor agonist very recently approved by the Food and Drug Administration (FDA) with indications for PH patients in World Health Organization functional class (WHO-FC) II and III (class B, level I) [15].
Many of the common side effects that were reported for prostanoids in single trials, e.g. flushing, headache, hypotension, nausea, vomiting and diarrhea, are generally dependent on the administered dose and they may disappear as treatment continues. Again, such reactions were reported for both parenteral and non-parenteral forms of treprostinil. Several symptoms can also arise as a consequence of vascular bed vasodilation by prostacyclin analogues and prostacyclin receptor agonists: the risk of events such as headache and flushing was increased by more than 1.5 fold. A two-fold increased risk for insomnia (treprostinil, p.o.) and 1.5 fold increased risk for cough (iloprost, inhaled) were also found. As demonstrated in a recent analysis by Leary et al. (2017) ADRs such as skin reactions, headaches and jaw pain were not associated with the mortality of patients receiving treprostinil (s.c.), while gastrointestinal side effects occurring during the first 8 weeks following treprostinil infusion were associated with a 57% increase in the risk of mortality. The authors attribute this phenomena to poor nutrition caused by gastric events; when accompanied with low albumin and body mass index, this can worsen prognosis in PH patients [7].
Another target for PH-specific agents is the endothelin pathway. ERAs play an essential role in the monotherapy of PH patients classified in WHO-FC II and III (class A, B-macitentan, level I), as well as being a component of both initial and sequential combined regimens [15]. The previous results from single clinical trials revealed that elevated liver aminotransferase values greater than three times normal occurred in 12.7% of patients receiving bosentan [16]. Twelve trials with an ERA therapeutic arm were included in the current meta-analysis, and this class of agents was found to be moderately safe. The most essential ERA-induced ADRs were classified among blood and lymphatic disorders with the subcategory anemia: iron deficiency is reported in 43% of patients with iPAH. Such events may be associated with reduced exercise capacity and with higher mortality, independent of the presence or severity of anemia [17]. The relative risk of anemia was elevated by more than one and half times during therapy. It was independent of PH-targeted therapy and it can be hypothesized that it was a consequence of clinical worsening.
Peripheral edema was demonstrated to be one of the most common adverse effects shared by prostanoid as well as ERA therapies in PH. It was suggested that the fluid retention induced by ERAs might be a consequence of both the primary vascular and renal effects of endothelin receptor type A blockade [18]. However, in our analysis, such events were demonstrated to significantly increase, independently of therapeutic class; the final outcome seemed to be closely determined by agents targeting the endothelin pathway.
As previously reviewed, treatment-related adverse events with PDE-5i are generally mild to moderate, and consistent for this class of therapeutic agents; this is an important fact from a clinical point of view. Due to potential benefit of the investigated PDE-5 inhibitors in PH patients, such as significant pulmonary vasodilation, current European guidelines strongly recommend them for WHO-FC II and III patients (class A, level I) as monotherapy or in combination with other PH-specific agents [17, 19]. The most common adverse events for PDE-5 inhibitors can include headache (46% vs. 39% placebo), flushing (12% vs. 4%), dyspepsia (12% vs. 7%), and back pain (12% vs. 11%) [19, 20]. The observations from earlier single trials performed on patients with erectile dysfunction are confirmed by those of our present survey, based on patients with PH. The risk of such events as headache or flushing was increased by more than one and a half times, and this seemed to be a consequence of the vasodilatory efficacy of sildenafil or tadalafil.
Eye disorders have been described previously as another adverse event specific to PDE-5 inhibitors and can involve such events as decrease in vision, flashes, bright colors, visual field defect, blurry vision, decrease in color vision or pain [21, 22]. In our survey, a substantial increase in relative risk of symptoms defined as ‘eye disorders’ was reported in cases where sildenafil but not tadalafil was added to the baseline. This phenomena can be explained by the 700-fold selectivity of tadalafil for PDE-5 over PDE-6.
Out of 36 trials that were included in the analysis, the participants of 18 received at least two PH-specific therapies. As we mentioned above, the combination regimens did not increase statistically significantly relative risk for severe ADRs. Conversely, other than severe ADRs were more pronounced in patients receiving the combined regimens. In relation to the particular events according to the MedRA dictionary, a significant tendency toward increased RR was observed for such ADRs as blood and lymphatic system disorders with anemia subgroup, gastrointestinal disorders with diarrhea and nausea subgroups, respiratory and thoracic diseases, nervous system disorders with headache, vascular events, myalgia and pain in limb. The first four main categories of ADRs tended to occur more often in the case of administration of at least two drugs as compared to monotherapy.
Our analysis has several limitations. First, it was not possible to obtain individual patient-level data from included trials, and this fact can weaken the accuracy of the obtained results. Second, of the 36 trials, only four concerned inhalation, two subcutaneous and one intravenous administration, which may have exaggerated the input of oral treatments on the results of safety. The third limitation concerned the variable reporting of adverse events (not all categories were included in all studies) and the different duration of trials, which could affect the observations of the safety of administered agents. Fourth, the definitions of severe ADRs were varied and commonly not provided in particular trials included in the meta-analysis. Fifth, due to the significant impact on the final outcome, some trials were removed during sensitivity analysis. This concerned only a few ADR categories. Similarly, our funnel plot analysis showed a graphic and statistical asymmetry for only a few ADRs. Nevertheless, publication bias favoring the publication of positive results (better safety profile) in such cases is possible. In addition, some of the reported side effects, such as dyspnea, fatigue, edema, anemia or cardiac disorders, may occur in response to underlying pulmonary hypertension. In this case, the interpretation of the ADRs associated with specific PH therapies can be inaccurate.
In conclusion, the applied therapies were associated with a non-significant risk of ADRs in more than half of their main categories according to MedRA. Conversely, the overall frequency for events classified into remaining categories (e.g. blood and lymphatic system, the gastrointestinal, musculoskeletal or nervous systems, or the respiratory or vascular disorders) were confirmed to be more than 1/10 or 1/100. Such ADRs as blood and lymphatic system disorders with anemia subgroup, gastrointestinal disorders with diarrhea and nausea subgroups, respiratory and thoracic diseases or nervous system disorders with headache were identified to occur more often in combination regimens as compared to monotherapy. Their risk can be increased when agents targeting prostacyclin pathway are used, especially. The risk of cessation was comparable between patients receiving PH-specific agents in monotherapy or combination, with the combined regimen demonstrating only a slightly greater risk.