Introduction
Traumatic brain injury (TBI) has become a major cause of death and disability worldwide in young individuals, and has received considerable concern in clinical practice [1]. Several studies have reported that TBI may even correlate with glioblastoma [2, 3]. In general, the injury of TBI can be divided into instantaneous primary mechanical damage and delayed secondary damage [4]. Primary damage is the insult which happens at the moment of impact including skull brain contusion, intracranial hemorrhages, diffuse axonal injury and skull fractures [5]. The secondary damage which has been proved may lead to many complications if left untreated, begins at the moment of injury and proceeds for hours or even days [6]. The hippocampus as well as the cerebral cortex in the brain may be affected and result in memory and spatial cognitive dysfunction after secondary damage following TBI [7]. Although several efforts such as control of brain edema, antibiotic treatment and surgical operation have been made, effective therapeutic strategies associated with neuronal death still remain uncertain [1].
Edaravone (3-methyl-1-phenyl-2-pyrazoline-5-one) is a kind of antipyrine derivative, which has been approved as the only potent free radical scavenger to treat acute cerebral infarction in the clinic since 2001 in Japan [8, 9]. Several studies have already indicated that edaravone was able to treat brain infarction, endothelial and oxidative damage as well as brain ischemia [10, 11]. Edaravone was also applied in other neuronal damage models including TBI and was discovered to attenuate oxidative stress levels and lesion size [12]. Furthermore, neuron apoptosis and neuronal degeneration induced by free radicals were inhibited and cerebral dysfunction was ameliorated with edaravone administration after TBI [13].
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophic factor family, which plays an important role in recovery and plasticity [14, 15] not only in the central nervous system, but also throughout peripheral nervous systems [16]. The expression level of BDNF has been demonstrated to be related to the decrease of neurons rose in a rat TBI model, which may have an impact on neuroprotective potential [17]. For the time being, some studies have found that the neuroprotective effects of edaravone following cerebral injury were correlated with the upregulation of BDNF [18, 19]. In addition, a recent study demonstrated that activation of the BDNF/TrkB signaling pathway inhibited neuronal cell apoptosis to perform its neuroprotective effect [20]. As a result, we speculate that edaravone may play its neuroprotective role through decreasing apoptotic neurocytes via the BDNF/TrkB signaling pathway. Nevertheless, although several studies and experiments mentioned previously provided insight into the mechanism of the neuroprotective function of edaravone, the real mechanism is still unclear and needs further study.
In the present study, we performed experiments to establish a rat TBI model and investigated the influence of TBI and edaravone on neurobehavior as well as learning and memory ability. We evaluated the expression of BDNF, TrkB and apoptosis related protein caspase-3, B-cell lymphoma-2 (Bcl-2), and Bcl-2 associated X protein (Bax), as well. We also probed apoptotic cells in the hippocampus caused by TBI and the effect of edaravone on neuronal apoptosis. Our study revealed the functional mechanism of edaravone on TBI associated with the BDNF/TrkB signaling pathway, which may provide a new therapeutic strategy of TBI in the clinic.
Material and methods
Animals and maintenance
A total of 75 adult healthy male Sprague-Dawley (SD) rats, weighing 200–240 g, were purchased from the Laboratory Animal Center of Kunming Medical University. All procedures in this experiment were approved by the Institutional Medical Experimental Animal Care Committee of the Affiliated Yantai Yuhuangding Hospital of Qingdao University. Animals were housed under controlled light conditions with a 12 h day and night cycle and supplied with food and water. Rats were placed in a warm condition to keep their body temperature stable.
Traumatic brain injury model
Following 10% chloral hydrate anesthesia (3 ml/kg), a midline longitudinal incision was performed to expose the skull between bregma and lambda suture lines. A steel disk (diameter 10 mm; thickness 3 mm) was adhered to the skull using dental acrylic. Animals were moved onto a foam blanket underneath a weight-drop device where a weight of 450 g fell freely through a vertical tube from 1.5 m onto the steel disk. Animals in the sham-operated group underwent the same surgical procedure as the TBI group without a weight-drop impact at the same time. Rats were then placed on heat pads at 37°C for 2–4 h to maintain normal body temperature during the recovery period.
Groups and drug administration
All rats were randomly separated into three groups as follows: sham group (n = 25), TBI group (n = 25), TBI + edaravone group (n = 25). Edaravone (Mitsubishi Pharma Corporation, Tokyo, Japan, dissolved in 0.9% saline solution) was injected intraperitoneally (i.p.) at a dose of 3 mg/kg at 30 min, 12 h, and 24 h after the TBI insult. Both sham and TBI groups received equal volumes of 0.9% saline by intraperitoneal injection at the same time. Each sub-group was composed of five rats and all researchers were blind and the animal codes were not revealed until the end of the histological and behavioral analyses.
Neurobehavioral evaluation
The severity of neurological deficit was assessed by the Neurological Severity Score (NSS) system by observers unaware of the treatment. Neurobehavioral function is an 18-point scale based on the presence of motoric, sensory, reflex, and balance (0, normal score; 18, maximal deficit score). The recovery of neurologic function was observed and the scores of all rats were recorded at 1, 4, 7, 14 and 21 days after treatment.
Morris water maze test
Variants of the MWM paradigm were used in order to evaluate spatial learning and memory of rats. The test was conducted at 5–8 days after injury, and each rat was tested for five trials per day for 4 consecutive days. The time required (escape latency) to find the hidden platform with a limit of 60 s was recorded by a video camera suspended above the maze together with a video tracking system (HVS Imaging, Hampton, UK). After that, the average escape time of a total of five trials was analyzed. On day 9 of the test, spent percentage of time in the target quadrant and swim speeds of the animals were investigated after the platform was removed.
Sample harvest
Ten days after injury, the rats were intraperitoneally anesthetized with 3.6% chloral hydrate (1 ml/100 g) and thoracotomy toward the direction of the cannula was performed to expose the heart. 400 ml of normal saline was infused and then fixed with 4% paraformaldehyde 500 ml. The brains were quickly removed and post-fixed with 4% paraformaldehyde. Then, the brains were dehydrated in 15%, 30% sucrose diluted with 4% paraformaldehyde. The injured hippocampus used for real-time polymerase chain reaction (RT-PCR) and western blot was only infused with normal saline and stored at –80°C.
Western blot
Total proteins were extracted from the hippocampus tissues, and protein concentration was assessed by the bicinchoninic acid (BCA) reagent (Thermo Fisher, Waltham, MA, USA) method. Equal amounts of protein were subjected to SDS-polyacrylamide gel electrophoresis. Separated proteins were transferred from the gel onto PVDF membranes (Bio-Rad, Hercules, CA, USA) by a transfer apparatus. The membrane was blocked with 5% fat-free dry milk for 2 h at room temperature. Subsequently, blots were incubated overnight at 4°C with indicated primary antibodies, including rabbit anti-caspase-3 (Sigma, St. Louis, MO, USA, 1 : 1000), rabbit anti-Bax (Sigma, 1 : 1000), rabbit anti-Bcl-2 (Sigma, 1 : 500) and rabbit anti-β-actin (Sigma, 1 : 1000). The membranes were then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Sigma, 1 : 5000) for 2 h at 37°C. Finally, immunoreactive bands of the protein expression level were normalized to the intensity of the corresponding bands for β-actin. Densitometric analysis of the results was performed with Image J 1.41 software (National Institutes of Health, Bethesda, MD, USA).
Real-time polymerase chain reaction
Total RNA was extracted from the injured cortex with Trizol Reagent (Invitrogen, Carlsbad, CA, USA) prior to cDNA synthesis. Reverse transcription to cDNA was executed according to the instructions of Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher). Detailed sequence information was as listed in Table I. PCR amplification was performed in a DNA thermal cycler (ABI 7300, Thermo Fisher) and conducted with the following standard protocol: one cycle of 94°C for 5 min; 35 cycles of 94°C for 1 min; annealing for 1 min and 7°C for 1 min. Relative gene expression was computed by the 2–ΔΔCt method.
Transferase-mediated dUTP-biotin nick end labeling assay
Transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining was executed with a TUNEL staining kit following the manufacturer’s protocol (Roche Inc., Basel, Switzerland). Sections of rat hippocampus were cultured with TUNEL reaction mixture containing TdT enzyme as well as fluorescein isothiocyanate (FITC) labeled dUTP in a dark and humid room for 1 h at 37°C, followed by a final wash for 15 min and visualized using a converter peroxidase (POD) with 4’,6-diamidino-2-phenylindole (DAPI). The number of TUNEL positive cells for five fields in the bilateral hippocampus of each section was counted under an Olympus BX43 fluorescence microscope (Olympus, Tokyo, Japan). An average for the five slices per brain was taken.
Flow cytometry assay
The flow cytometry assay was performed with the Annexin V-EGFP apoptosis detection kit (Beyotime, Jiangsu, China). Cells were collected in Eppendorf (EP) tubes after cutting, grinding and suspending. Then the cells were washed with phosphate buffered solution (PBS) and resuspended in 500 μl of binding buffer. After that, 5 μl of Annexin V-FITC and propidium iodide (PI) solutions were added to the cells, which were incubated subsequently for 15 min on ice. Finally, the apoptosis rate was analyzed by coulter FC500 flow cytometry (Beckman Coulter, Miami, FL, USA).
Results
Edaravone improved neurobehavior in rats following TBI
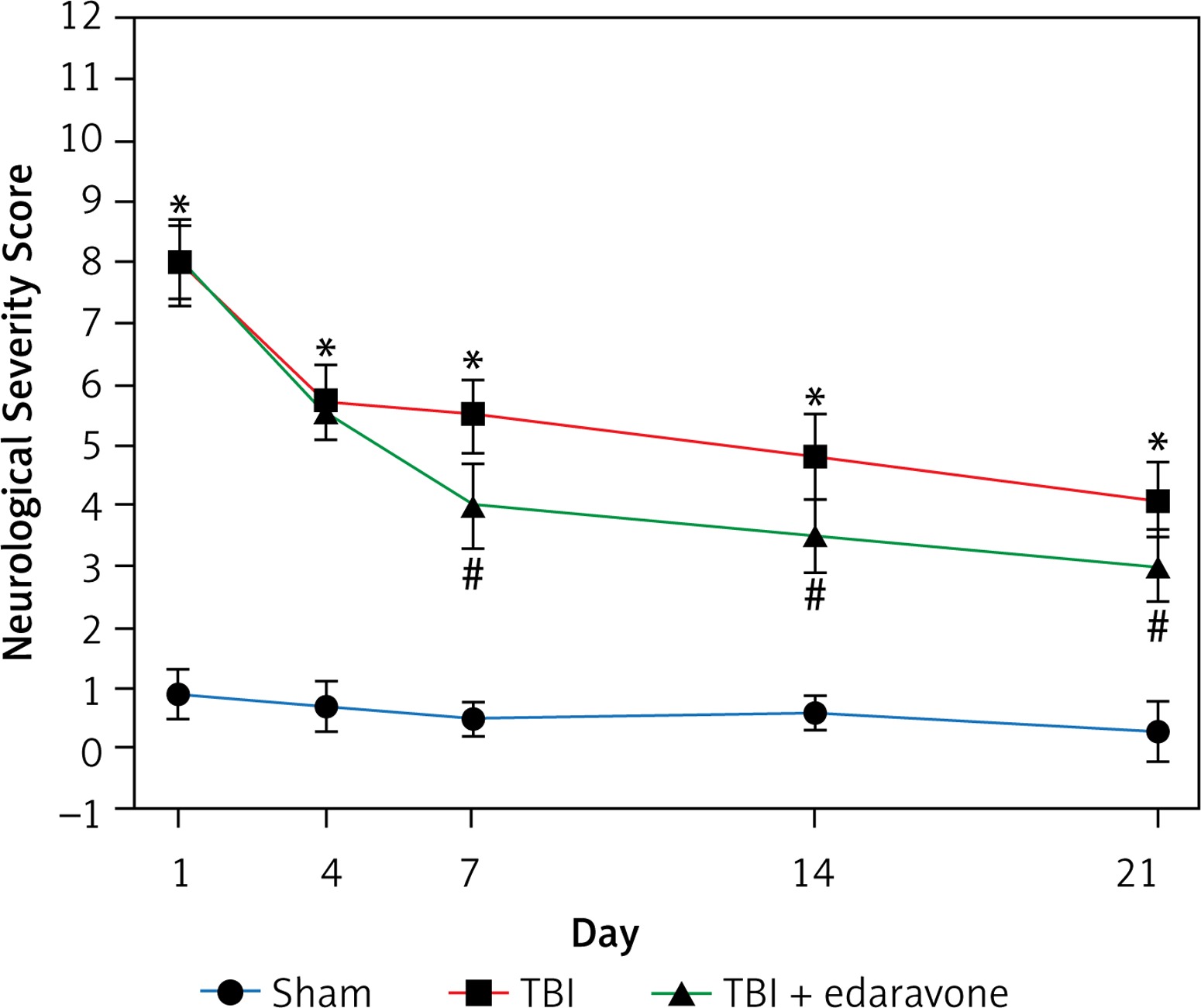
The NSS score was assessed at 1, 4, 7, 14 and 21 days following cerebral injury. At each time point detected in the experiment, the NSS score in the TBI group was significantly higher than that in the sham group (p < 0.05). In the group with edaravone administration, the NSS score was significantly decreased at 7, 14, 21 days compared with the TBI group (p < 0.05, Figure 1). The promotion in neurobehavior showed that edaravone exerted a neuroprotective effect.
Figure 1
Effect of edaravone on neurobehavior. The Neurological Severity Score (NSS) of rats was significantly increased in the traumatic brain injury (TBI) group. Administration of edaravone significantly improved motor function at 7–21 days, as reflected by a decrease in NSS
*p < 0.05, compared with sham group; #p < 0.05, compared with TBI group.
Edaravone improved the spatial memory and learning ability
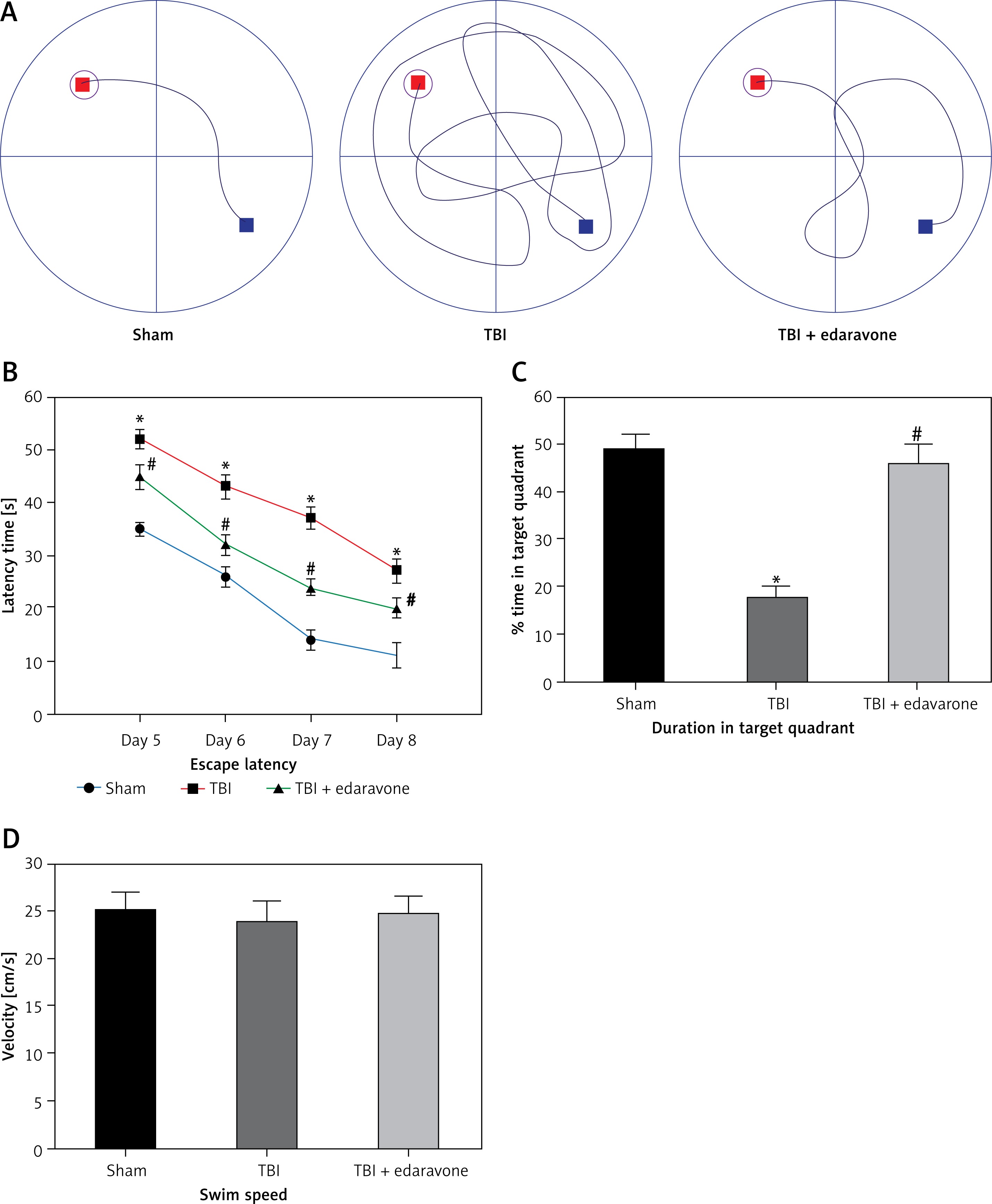
After 20 trials over a period of 4 days, animals in the TBI group exhibited significant spatial memory deficits compared with the sham group (p < 0.05). Furthermore, the edaravone administration group performed obviously better than the TBI group in a few parameters, commonly used to evaluate the ability of hippocampal-dependent spatial memory and learning (p < 0.05), such as escape latency (Figures 2 A, B) and time spent in the target quadrant (Figure 2 C). However, there were few differences in swim speed among these groups (p > 0.05, Figure 2 D), indicating that the observed differences of escape latency were not a consequence of an inability to execute the swim task. The results suggested that edaravone promoted spatial memory and cognitive ability outcome following TBI.
Figure 2
Effect of edaravone on learning and memory ability. A – Swimming trajectories of each group on day 8. B – The escape latency increased remarkably at 5, 6, 7, and 8 days following TBI. C – On day 9, sham and TBI + edaravone groups spent a greater percentage of time in the target quadrant compared with TBI group. D – There were no significant differences in swim speeds among groups
*p < 0.05, compared with sham group; #p < 0.05, compared with TBI group.
Edaravone increased BDNF and TrkB level in TBI rat brain
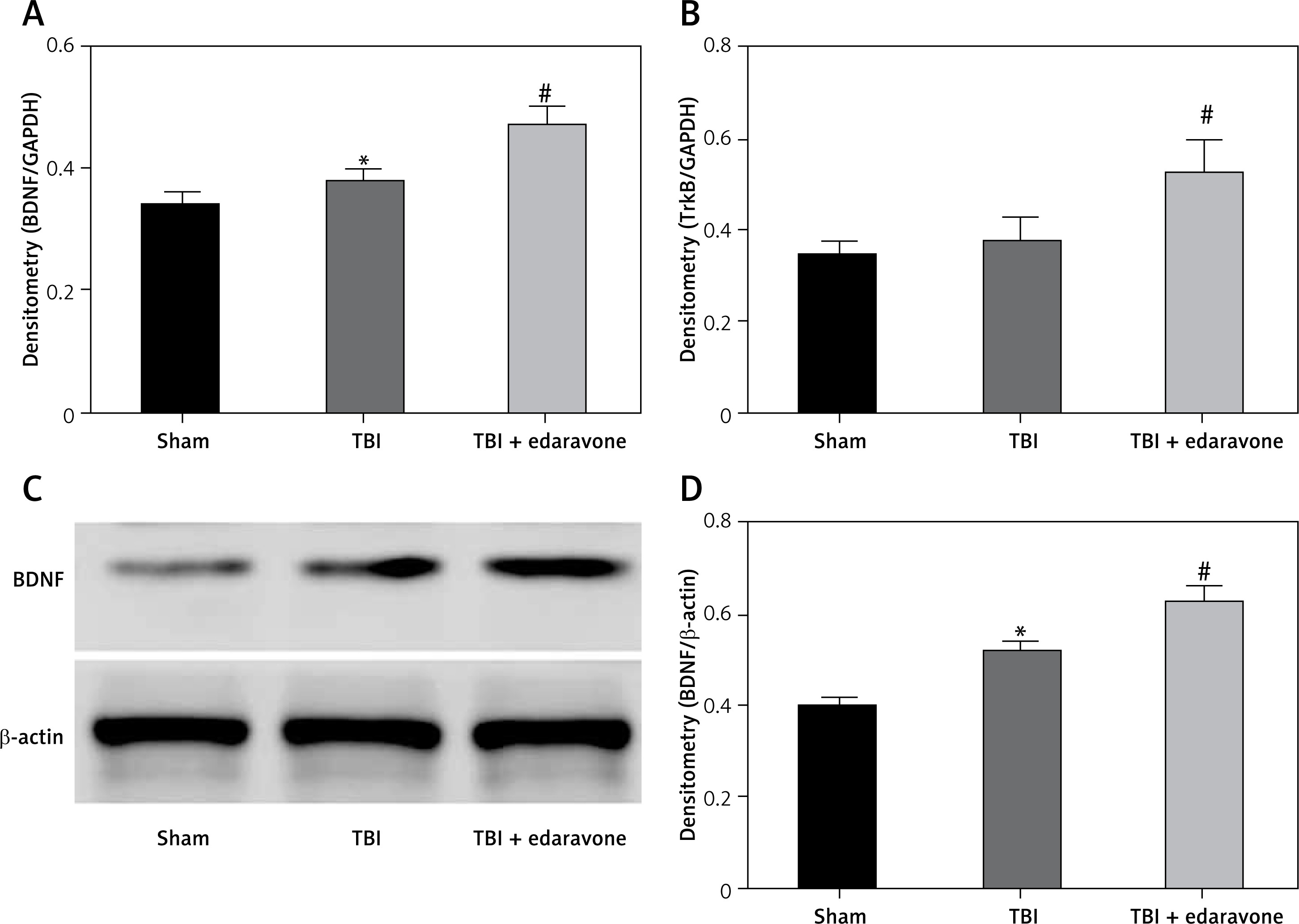
Through RT-PCR, the BDNF and TrkB levels were low in the sham group and significantly increased at 48 h after TBI (p < 0.05), and BDNF and TrkB levels were significantly higher in the TBI + edaravone group compared with the TBI group (p < 0.05, Figures 3 A, B). In addition, western blot indicated the same result of the expression of BDNF as RT-PCR (Figures 3 C, D), which suggested that edaravone treatment upregulated BDNF and TrkB levels after TBI.
Figure 3
Effect of edaravone on expression of brain-derived neurotrophic factor (BDNF) and tyrosine kinase receptor B (TrkB). A – Reverse transcription-polymerase chain reaction (RT-PCR) indicated that BDNF mRNA level rose with edaravone treatment following TBI compared with sham group. B – Similarly, compared with sham group, expression of TrkB mRNA was enhanced after edaravone. C – Western blot analysis demonstrates levels of BDNF in the hippocampus of rats at 48 h. D – The result of western blot demonstrated that BDNF level was upregulated by edaravone
*p < 0.05, compared with sham group; #p < 0.05, compared with TBI group.
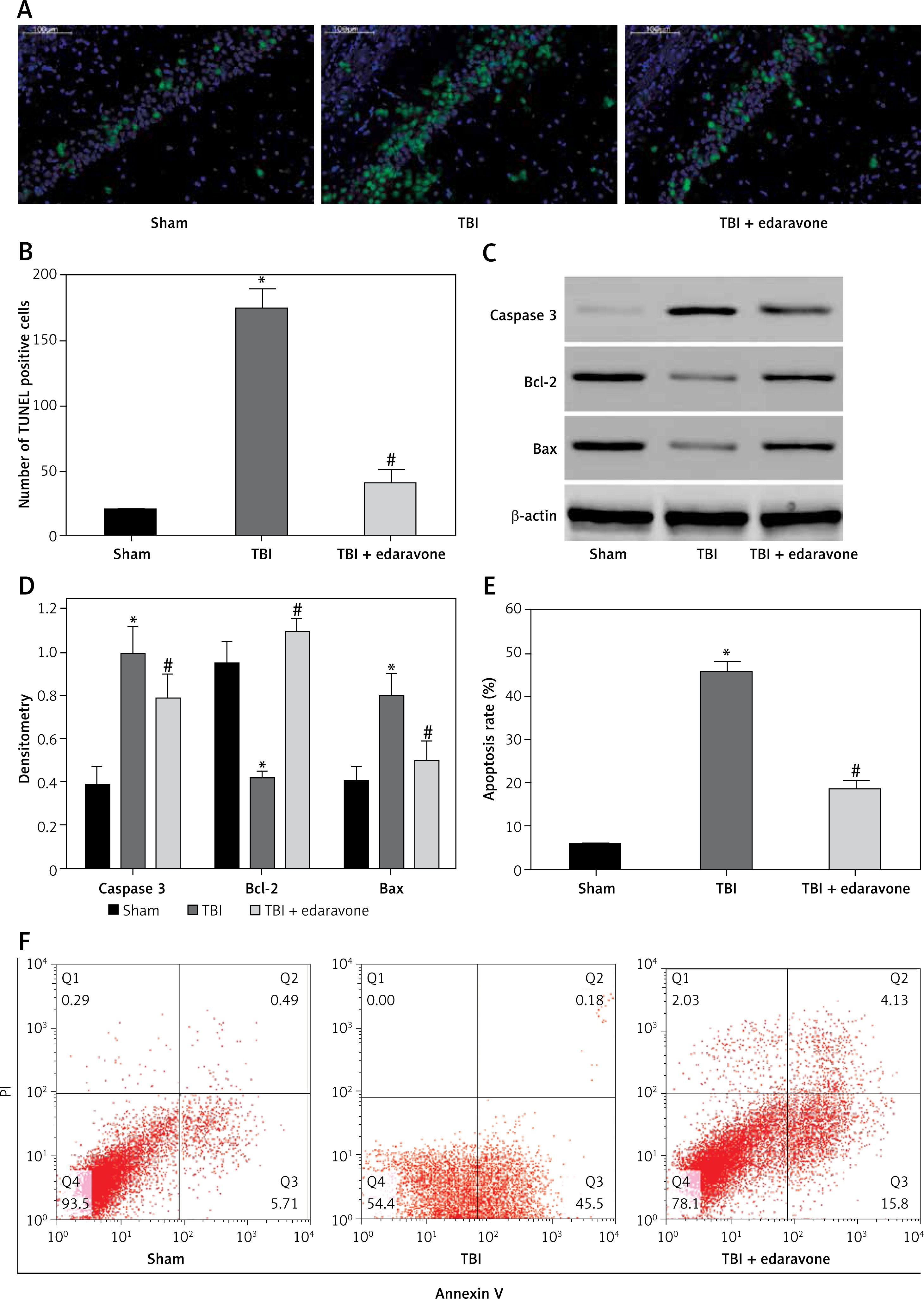
Edaravone inhibited neuronal apoptosis following TBI
Through TUNEL staining assay, a significant increase in TUNEL positive cells at 48 h in the rat hippocampus of the TBI group was observed compared with the sham group (p < 0.05). Edaravone treatment notably reduced the amount of TUNEL positive cells compared to the TBI group (p < 0.05, Figures 4 A, B), which represented a lower apoptosis rate. According to the results of western blotting with caspase-3, Bcl-2, and Bax in the hippocampus of rats at 48 h (Figure 4 C), treatment with edaravone significantly reduced the protein expression of caspase-3 (p < 0.05). Compared with the sham rats, the protein expression of Bcl-2 decreased, whereas Bax increased in TBI rats (p < 0.05). Treatment with edaravone remarkably upregulated the protein expression of Bcl-2 and decreased the protein expression of Bax (p < 0.05, Figure 4 D). Flow cytometry assay was performed to evaluate the apoptosis rate. The cell apoptosis rate in the hippocampus was significantly increased after TBI (p < 0.05), while edaravone administration inhibited cell apoptosis dramatically in the rat TBI model (p < 0.05, Figures 4 E, F). These results strongly suggested that edaravone attenuates neuronal apoptosis in the rat TBI model.
Figure 4
Edaravone inhibited neuronal apoptosis in hippocampus. A – Representative images of TUNEL staining in hippocampus from each group at 48 h (100 μm). Transferase-mediated dUTP-biotin nick end labeling (TUNEL) positive cells displayed green staining within the nucleus of apoptotic cells. B – Through TUNEL assay, TBI induced neuronal apoptosis which could be attenuated by edaravone treatment. C – Western blot analysis demonstrated levels of caspase-3, B-cell lymphoma-2 (Bcl-2), Bcl-2 associated X protein (Bax) in hippocampus of rats at 48 h. D – The result of western blot indicated that expression of caspase-3 and Bax in TBI+edaravone was lower than that in TBI group. However, Bcl-2 level showed the opposite trend. E, F – Flow cytometry assay revealed that neuronal apoptosis rate was decreased in TBI + edaravone group compared with TBI group
*p < 0.05, compared with sham group; #p < 0.05, compared with TBI group.
Discussion
In the present study, we found that neurological functional outcome such as memory and spatial cognition was promoted significantly with edaravone treatment following TBI insult. The expression of BDNF and TrkB was enhanced following edaravone administration in a rat TBI model. In addition, the neuronal apoptosis rate declined and the expression of apoptosis related protein caspase-3 and Bax was decreased, while that of Bcl-2 was upregulated with edaravone treatment in the hippocampus. The results of our study revealed that edaravone attenuates neuronal apoptosis via activation of the BDNF/TrkB signaling pathway.
Traumatic brain injury has already become a public health concern all over the world. Patients with TBI suffer from neuropsychiatric and physiological impairments including depression, memory and cognitive dysfunction [21]. Edaravone is regarded as a typical free radical scavenger which is used as a neuroprotective agent against acute cerebral injury in the clinic [22]. Ohta et al. found that edaravone protected against cognitive impairments after TBI via attenuation of oxidative stress and axonal injury [23]. In another study, Lee et al. suggested that the neuroprotective effect of edaravone also came into effect in vitro [24]. Furthermore, Jiang et al. investigated the effect of edaravone compared with ginsenoside Rb1 and found that the therapeutic effect of ginsenoside Rb1 might be mediated by decreasing inflammation and promoting nerve cell growth, while that of edaravone is correlated with its antioxidant function [25]. Similarly, our study reported that edaravone administration improved neurobehavior, memory and spatial cognition outcome following TBI.
BDNF is a representative neurotrophic factor in the brain, playing a neuroprotective role after cerebral injury, and has been proved to be vital to brain injury [26, 27]. For instance, Satoshi et al. found that the neuroprotective effects of edaravone in brain ischemia were correlated with the upregulation of BDNF expression, which may activate the downstream signaling pathway [18]. Similarly, Yuan et al. found that the upregulation of BDNF with edaravone administration protected against retinal damage caused by diabetes, as well [28]. Our study came to the same conclusion that edaravone was able to enhance the expression level of BDNF and TrkB, as well, which suggested that the neuroprotective function of edaravone came into effect through the induction of BDNF production.
Apoptosis is crucial for tissue homeostasis and embryogenesis, and its deregulation leads to autoimmune disorders, immunodeficiency, or cancer [29, 30]. Neuronal apoptosis is a main contributor to not only primary brain damage, but also secondary brain damage after TBI insult, which determines the outcome of TBI patients [31]. Kim et al. reported that the increase of neurotrophic factors including BDNF via mesenchymal stem cells led to the decline of the number of apoptotic nerve cells following TBI [1, 4–28, 31–34]. Wu et al. found that 7,8-dihydroxyflavone (DHF), a TrkB receptor agonist, inhibited neuronal apoptosis via PI3K/Akt signaling [20]. At the same time, Alder et al. reached a similar conclusion and found that p75 had the opposite effect to TrkB [33]. In addition, our study confirmed that a high level of BDNF related to low neuronal apoptosis after TBI was still valid in a murine TBI model [34]. All these cited papers support our viewpoint that edaravone attenuates neuronal apoptosis following TBI through the BDNF/TrkB signaling pathway.
Our study indicated that the neuroprotective effect of edaravone was targeted at the BDNF/TrkB signaling pathway, which may provide a new clinical therapeutic strategy of TBI. Nevertheless, there were still some limitations in the study which ought to be taken into consideration. For instance, only one dose of edaravone was used in this experiment and only apoptotic cells in the hippocampus were investigated. As a result, the effect of edaravone on TBI in different doses and time windows as well as apoptotic nerve cells in the cerebral cortex requires further study.
In conclusion, the results of our study provided experimental evidence for the application of edaravone in the treatment of TBI. The neuroprotective effects of edaravone such as attenuating neuronal apoptosis were related to activation of the BDNF/TrkB signaling pathway.