Introduction
Osteoarthritis (OA) is a chronic disease which features loss of cartilage and joint pain associated with inflammation [1, 2]. OA is associated with alterations in subchondral bone, degeneration of cartilage and inflammation of the synovial membrane [3]. Literature suggests that none of the present therapeutic approaches can halt the progression of the disease successfully. The majority of treatments for OA are focused on controlling the symptoms, including pain, and involve use of drugs belonging to the non-steroidal anti-inflammatory (NSAIDs) category, or paracetamol is the choice in some cases [4, 5]. However, long-term use of these drugs leads to severe GI disturbances, and adverse hepatic and cardiac events; hence there is an urgent need for agents which provide a safe and effective route for treating OA. An agent from food-derived sources (nutraceuticals) would be more advantageous [6, 7].
Enoxolone (EN), also called glycyrrhetinic acid, is a pentacyclic triterpenoid obtained from the herb liquorice. EN is used as a flavoring agent in food and is also used to mask the bitter taste of drugs such as aloe and quinine. EN has been discovered to show numerous pharmacological activities including anti-inflammatory, anti-oxidant, gastroprotective, antiviral, cardioprotective, anti-tumor, neuroprotective and hepatoprotective activities in animal models [8–10]. EN is reported to exert a potent anti-inflammatory effect by reducing inflammatory edema in an animal model of myocardial damage [11]. EN has also been found to modulate vascular injury and atherogenesis [12]. Since osteoarthritis is associated with inflammatory conditions of synovial joints, we postulated that EN can be a potential candidate for treating osteoarthritis. Interleukin 1β (IL-1β) is a pro-inflammatory cytokine that has been reported to play a vital role in the pathogenesis and factors contributing to osteoarthritis. Upon release IL-1β induces the synthesis of some more pro-inflammatory cytokines which are responsible for stimulation of apoptosis in chondrocytes [13]. Cytokines are reported to be potent stimulators for generation of catabolic enzymes such as matrix metalloproteinases (MMPs). MMPs are markers indicating excessive degradation of matrix in osteoarthritis [14]. Interleukin 1β is also responsible for suppression of cartilage-specific proteoglycans, extracellular matrix and collagen [15]. These changes in catabolism can provide acceleration of degeneration of cartilage. In the present study we analyzed whether EN could inhibit apoptotic activity of IL-1β.
Autophagy is a conserved catabolic process; it leads to degradation of non-functional organelles and damaged proteins [16–18]. Also, autophagy exerts a protective effect in the process of degeneration of fibro-chondrocytes and chondrocytes [19, 20]. Autophagy decreases with increasing age, which may be a factor contributing to osteoarthritis in elderly peoples, and hence activation of autophagy is very important for reducing the severity of OA [21]. EN has been reported to induce autophagy in hepatocellular carcinoma [22], but the role of EN in autophagy of chondrocytes has not been studied. In the present study we evaluated the role of EN in chondrocyte autophagy.
A number of studies have suggested involvement of signaling pathways responsible for malfunctioning in chondrocytes and synovial cells causing aging, rheumatoid arthritis and osteoarthritis. A study confirmed that extracellular signal-regulated kinase 1/2 (ERK1/2) is associated with apoptosis in chondrocytes [23]. There is a growing need for novel agents for treating osteoarthritis which can target chondrocytes via multiple cellular pathways by suppressing inflammation without any side effects. Recently it has been proved that EN can modulate inflammation via the mitogen-activated protein kinase (MAPK) pathway [24]. EN has also been found to target the extracellular signal-regulated kinase (ERK) pathway [25]. Both MAPK and ERK are required for survival, differentiation and maintenance of chondrocytes [23]. The aim of the present study was to evaluate any link between enoxolone and autophagy in IL-1β stimulated Iry chondrocytes in vitro, and if there is a correlation, what pathway is involved.
Material and methods
Animals
For the study we used male Sprague-Dawley rats weighing 210–220 g, each group comprising 6 animals. The animals were obtained from the animal center of Beijing Chaoyang hospital, Capital Medical University, Beijing China. All the experiments were approved by the Animal Ethical Review Board of Beijing Chaoyang Hospital, China. The ethical approval number was AJ001457. The animals were housed under controlled conditions of temperature (25°C) in a laminar flow in a 12 h dark and light cycle. The animals were provided with free access to food and water. The animals were subjected to isoflurane anesthesia (2.5%) as required. All the animals were sacrificed at the end of the study by cervical dislocation by a trained individual.
Reagents
Enoxolone, collagenase-II, rapamycin and Dulbecco’s Modified Eagle Medium F-12 (DMEM/F12) were procured from Sigma-Aldrich USA. Bovine serum albumin (BSA) and fetal bovine serum (FBS) were obtained from Thermo-Fisher, USA. Methyladenine (PI3K inhibitor) was procured from InvivoGen USA, and was used in a solution for 100 mM by dissolving in phosphate buffer saline (PBS). IL-1β, Terminal deoxynucleotidyl transferase dUTP nick end labeling and apoptosis assay kits were bought from Abcam USA. The antibodies used for the study were anti-cleaved caspase-3 (1 : 1000), anti-beclin-1 (1 : 1000), anti-Bcl-2 (1 : 1000), anti-LC-3 (1 : 1000), anti-ERK (1 : 1000), anti-GAPDH (1 : 1000) and anti-ERK (1 : 1000), which were bought from Cell Signaling Technology, USA. The anti-TAMRA and anti-collagen antibody were supplied by Abcam USA. DAPI and Alexa Fluor 488 Phalloidin were purchased from KeyGen Biotech China. The Cell Counting Assay Kit-8 was supplied by InvivoGen USA.
Isolation, culture and identification of chondrocytes
The cartilage tissue samples were collected from femoral head articular cartilage of healthy SD rats aged 4 weeks. The tissues were trypsinized enzymatically using trypsin reagent (0.25%) along with EDTA and collagenase-2 (0.25%), maintaining a temperature of 37ºC and 5% CO2 for 5 h. The Iry chondrocytes (1 × 105 cells/cm2) were transferred to petri-dishes having high density monolayers; the culture media consisted of DMEM/F12 (50 : 50) supplemented with FBS (10%), ascorbic acid (25 mg/ml), streptomycin (50 IU/ml), penicillin (50 IU/ml), essential amino acids, amphotericin B (2.5 mg/ml) and L-glutamine. For all the experiments freshly obtained Iry chondrocytes were obtained and used.
The chondrocytes were identified by immunofluorescence staining. The cells were subjected to seeding in chamber slides. The obtained chondrocytes were rinsed with phosphate buffer saline at least twice and were fixed with paraformaldehyde (4%) for 15 min, permeabilized using Triton X-100 (0.5%) for 5 min and were blocked finally with bovine serum albumin (1%) for 10 min. The chondrocytes were incubated along with anti-collagen-2 Iry antibody for 60 min, then washed twice with PBS and again incubated with TRITC conjugated IIry antibody for 60 min. The nuclei and the cytoskeleton were stained with DAPI and phalloidin for 10 min. The obtained fluorescent images were analyzed with Olympus Software (Olympus, Japan).
Cell viability studies and cell proliferation assay
The isolated and confirmed chondrocytes were used for cell viability and proliferation studies. The cell viability was done by Cell Counting Assay Kit-8 as per the provided instructions. Briefly, 5 × 103 cells/well were cultured in a 96-well plate for 24 h, after which the control cells (untreated) received IL-1β (10 ng/ml) alone for defined time periods, the cells receiving pre-treatment with EN (10 µM) or rapamycin (7.5 µM) for 4 h, followed by treatment with IL-1β (10 ng/ml), EN (10 µM) or rapamycin (7.5 µM) for defined time intervals. The experimental dilutions of EN (i.e. 5 µM, 10 µM, 15 µM and 20 µM) were prepared in the culture media (DMEM/F12 media as described earlier) added to predefined wells. In the same way, dilutions for IL-1β (10 ng/ml, 15 ng/ml, 20 ng/ml and 25 ng/ml) were prepared and added to culture media. As the dilutions of EN were done using DMSO, a control group was created by treating the cells with DMSO only. The cytotoxicity assay was done after 12, 24 and 48 h, and absorbances for the experiment were recorded on a UV-visible spectrophotometer (Shimadzu-1800, Japan) at 450 nm three times. The percentage of viable cells was calculated using the formula % cell viability = (optical density of treated/optical density of control) × 100.
TUNEL staining
For identifying apoptotic cells, TUNEL assay was done to find the fragmentation of DNA. For TUNEL assay, 1 × 106 cells/well chondrocytes were transferred to a 96-well plate and were treated with reagents, followed by incubation at room temperature. The cells were fixed using paraformaldehyde (4%) for 20 min at 37ºC, followed by washing twice with PBS and permeabilized using triton-x 100 (0.1%) and again washed twice with PBS. The apoptotic chondrocytes were subjected to staining using the cell death detection kit for TUNEL for in vitro studies. The cell nuclei were counterstained with DAPI. After that, the slides were mounted with cover-slips followed by observation under a confocal microscope for identifying apoptosis. The percentage of apoptotic cells was evaluated as the number of fluorescein labeled cells/DAPI-stained nuclei. For calculations 100 cells from 25 different microscopic fields were selected to confirm the number of cells with apoptotic morphology.
Autophagy by monodansylcadaverine assay
A monodansylcadaverine (MDC) assay kit (Abcam USA) was used to study autophagy in chondrocytes. MDC becomes incorporated with autophagic lysosomes emitting fluorescence present in the cytoplasm. The chondrocytes were cultured on sterile glass slides in the culture media. The Iry chondrocytes were either untreated or treated with EN (10 µM) or rapamycin (7.5 µM) for 2 h followed by treatment with IL-1β (10 ng/ml) and 3-methyl adenine (10 mM) for predefined time periods. After these treatments, the chondrocytes were incubated with monodansylcadaverine (0.05 mM) for 25 min at 37ºC. After incubation, the chondrocytes were rinsed three times using PBS and mounted on glass slides followed by coverslips, then the slides were viewed under confocal microscope. The excitation and emission wavelengths were 360 and 380 nm respectively; the images were analyzed using Olympus software (Olympus, Japan). The cells with autophagy morphology were identified and calculated, at least 100 cells/identified field were taken into account from 20 different sites in a slide.
Transmission electron microscopy (TEM)
About 1 × 106 cells were fixed with paraformaldehyde-glutaraldehyde solution (Karnovsky fixative) which comprised 20 ml of paraformaldehyde solution (16%), 8 ml of glutaraldehyde EM grade (50%), 25 ml of sodium phosphate buffer (0.2 M) and 25 ml of distilled water. After fixation, the cells were exposed to OsO4 (1%) (0.1 M phosphate buffer). The monolayer cell pellets were washed and dehydrated with ethanol. The dehydrated cells were embedded in Epon mixture and cut using a diamond knife using a Reichert-Jung Ultracut-E ultramicrotome (Reichert-Jung, Germany). The obtained ultrathin sections were stained with uranyl acetate/lead citrate followed by transmission electron microscope study using a TEM-10 microscope (Zeiss, Germany).
Western blot analysis
The expression of proteins was assessed by western blot analysis. To evaluate the effect of EN on IL-1β mediated apoptosis, the Iry chondrocytes were subjected to lysis using a radio-immunoprecipitation assay buffer (RIPA), to which was added a protease inhibitor cocktail followed by centrifugation at 10 000 rpm for 10 min to settle the cell debris. The total protein from cytoplasmic extracts and whole cell samples was estimated by a protein assay kit (Thermo Fisher USA) following the supplied instructions. Briefly, 25 µg of total protein in each lane was subjected to separation using 10, 12.5 and 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were subjected to blotting on a polyvinylidene fluoride (PVDF) membrane and were blocked using skimmed milk (2.5%) for 60 min at 37ºC. The membranes were incubated with Iry antibodies against Bcl-2, LC-3, p-ERK, ERK, Beclin-1 and GAPDH for 12 h at 4ºC. The membranes were rinsed three times using a mixture of tris-buffered saline (TBS) and polysorbate 20 called TTBS followed by incubation with IIry antibody for 60 min. The membranes were again washed by TTBS followed by densitometry analysis of each band using ChemiDoc Images (Bio-Rad, USA).
Flow cytometry analysis
After receiving the defined treatments, the chondrocytes were rinsed using ice-cold phosphate buffer saline and the cells were trypsinized in absence of EDTA using a trypsinizing reagent (Thermo Fisher USA). The supernatant was removed by centrifugation (10 000 rpm, 10 min), about 1 × 105 cells were suspended in binding buffer (200 µl) followed by incubation with Annexin V-FITC (4 µl) for 10 min at 37ºC in the dark. After this, propidium iodide (4 µl) along with binding buffer (300 µl) was added to the flow tube. The % apoptosis was evaluated using a flow cytometer (Invitrogen USA).
Ethics approval and consent to participate
For the study, male Sprague-Dawley rats were obtained from the animal center of Beijing Chaoyang Hospital, Capital Medical University, Beijing China. All the experiments were approved by the Animal Ethical Review Board of Beijing Chaoyang Hospital, China. The ethical approval number was AJ001457.
Statistical analysis
All the experiments were performed in triplicate; the data are presented as mean ± %RSD. The statistical analysis was done using GraphPad Prism software. Unpaired one-way ANOVA was done for more than two groups and the t test for analyzing data presented in two groups. P-values < 0.05 were considered as significant.
Results
Confirmation of chondrocytes by immunofluorescence (collagen II expression)
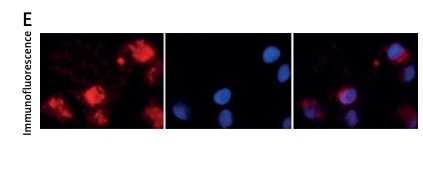
Collagen-2 is found to contribute 85–90% of the total collagen synthesized by chondrocytes in cartilage or primary culture [26–28]. Hence confirmation of chondrocytes was done by identifying the presence of collagen-2. Immunofluorescence staining of collagen-2 was done to identify the cells as chondrocytes. Red staining was seen in images of cell cultures, confirming the presence of collagen-2, which indicated that the isolated cells were Iry chondrocytes.
Effect of enoxolone and IL-1β on viability of chondrocytes
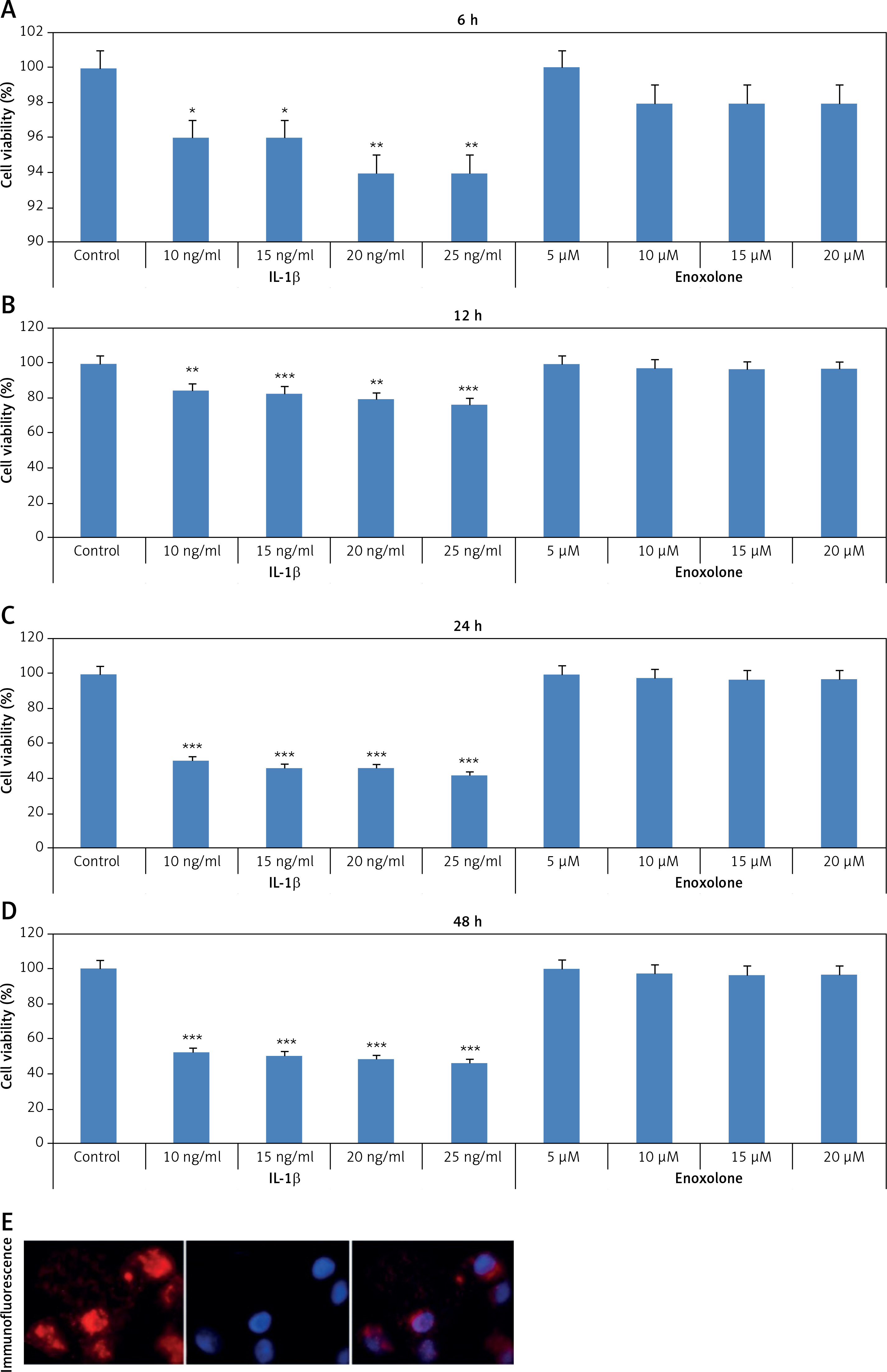
The effect of EN and IL-1β on cell viability was evaluated at various time intervals; the results are presented in Figures 1 A–E. The cytotoxicity of EN was evaluated by Cell Counting Assay Kit-8. The results suggested that EN showed no significant cytotoxic effect up to the concentration of 20 µM at the different time intervals (6, 12, 24, 48 h). However, the chondrocytes treated with IL-1β at all concentrations (10, 15, 20 and 25 ng/ml) showed a significant decrease in cell viability at the selected time intervals. The results suggested that treatment with IL-1β caused a significant decrease in cell viability of Iry neonatal chondrocytes with increasing dose and exposure time.
Figure 1
Effect of treatment with EN and interleukin 1β (IL-1β) on proliferation and viability of Iry chondrocytes in vitro. The cells were exposed to EN or IL-1β to study the cytotoxicity, 5 μM, 10 μM, 15 μM and 20 μM of EN or 10 ng/ml, 15 ng/ml, 20 ng/ml and 25 ng/ml of IL-1β for time intervals ranging from 6 to 48 h (A–D), where it was observed that IL-1β exerted significant cytotoxicity. The results are presented as mean ± SD. *P < 0.05, **p < 0.01 and ***p < 0.001 against controls. E – Identification of collagen II by immunofluorescence in Iry chondrocytes
Enoxolone inhibits the IL-1β induced apoptosis of chondrocytes
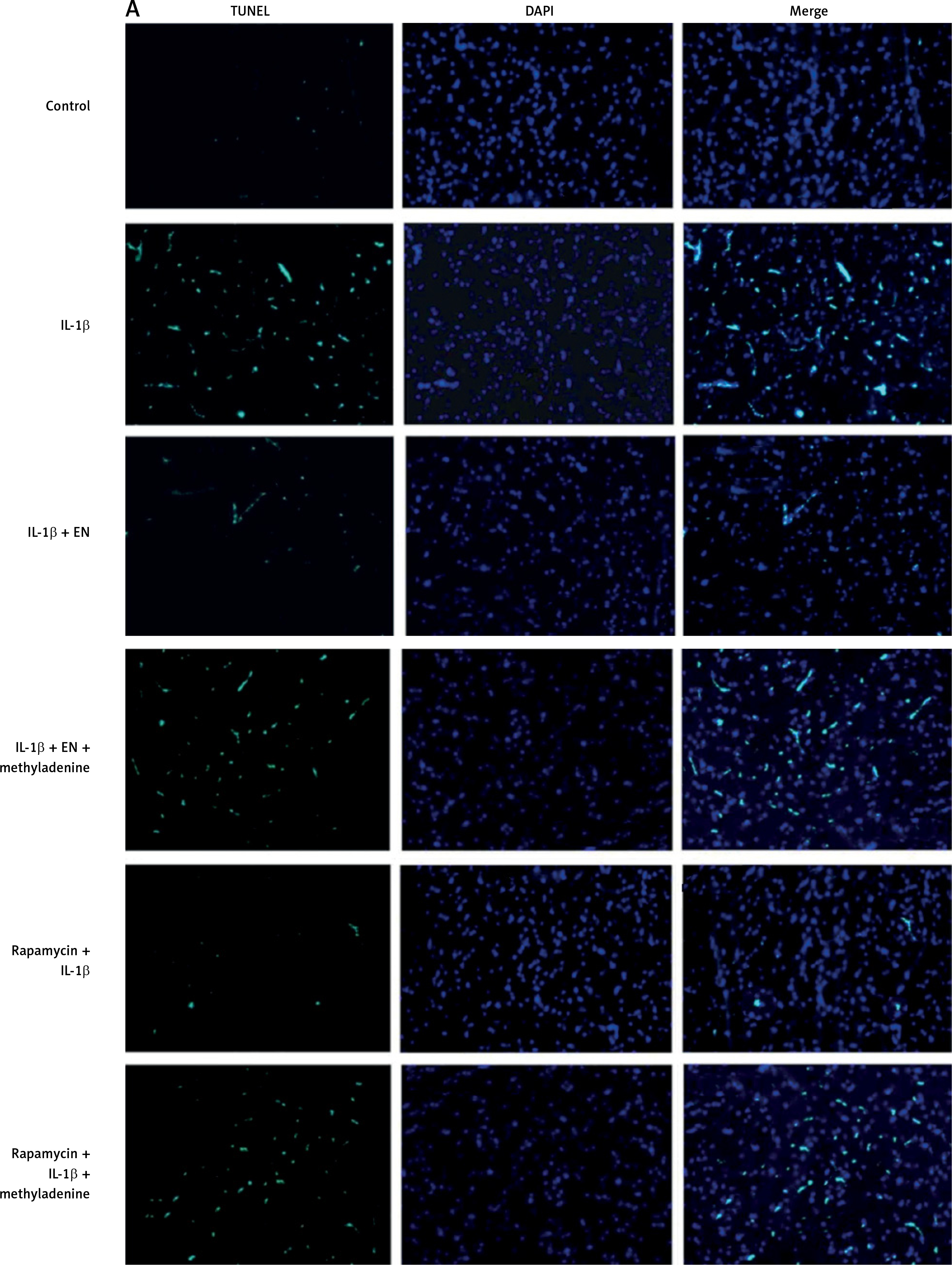
TUNEL staining and flow cytometric analysis was done to study the effect of enoxolone on IL-1β mediated apoptosis of chondrocytes. The serum starved isolated Iry chondrocytes were treated with IL-1β or EN + IL-1β or EN + IL-1β + 3-methyladenine or rapamycin + IL-1β or rapamycin + IL-1β + 3-methyladenine; the cells treated with vehicle served as a control. Apoptosis was analyzed by flow cytometry analysis and TUNEL assay. TUNEL staining is shown by cells existing in early stages of apoptosis as they contain nicked DNA. As shown, the chondrocytes receiving treatment with vehicle (Control) exhibited negative TUNEL staining, whereas chondrocytes treated with IL-1β showed a high extent of apoptosis with a significantly higher number of TUNEL positive cells (Figure 2 A) compared to those receiving treatment with EN + IL-1β. The TUNEL positive cells were again on the higher side in the group receiving treatment with EN + IL-1β + 3-methyladenine. Treatment with rapamycin along with IL-1β resulted in a significant reduction in TUNEL positive cells whereas when 3-methyladenine was added to this combination it resulted in increased TUNEL positive cells.
Figure 2
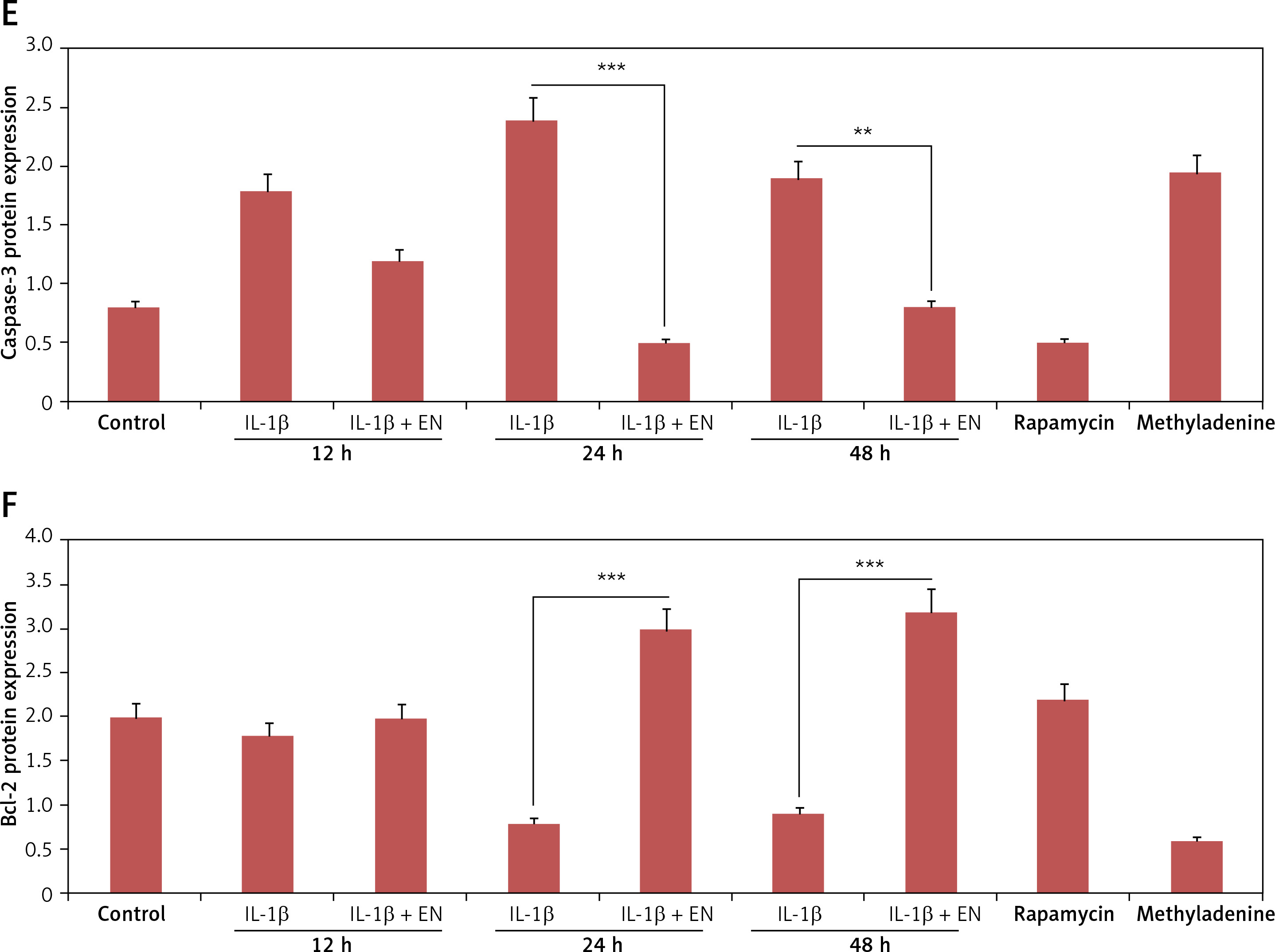
Effect of EN on IL-1β-mediated apoptosis in Iry chondrocytes. A – Terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) assay (Green) was done for visualization of apoptotic cells. 4′,6-diamidino-2-phenylindole (DAPI) staining (Blue) for nuclei. B – Indicates % TUNEL positive cells. C – Apoptosis study by flow cytometry was done in chondrocytes. The cells were labeled with Annexin V and propidium iodide and were analyzed by flow cytometry. The percentage of apoptotic cells in each quadrant is shown; the upper left shows necrotic cells, the lower left shows living cells, the lower right indicates early apoptotic cells and the upper right quadrant shows late apoptosis cells. D–F – Expression of caspase-3 and Bcl-2 by western blot analysis followed by various combinations of treatments comprising EN, rapamycin, methyladenine and IL-1β
Results are presented as means ± SD.*P < 0.05, **p < 0.01, ***p < 0.001.
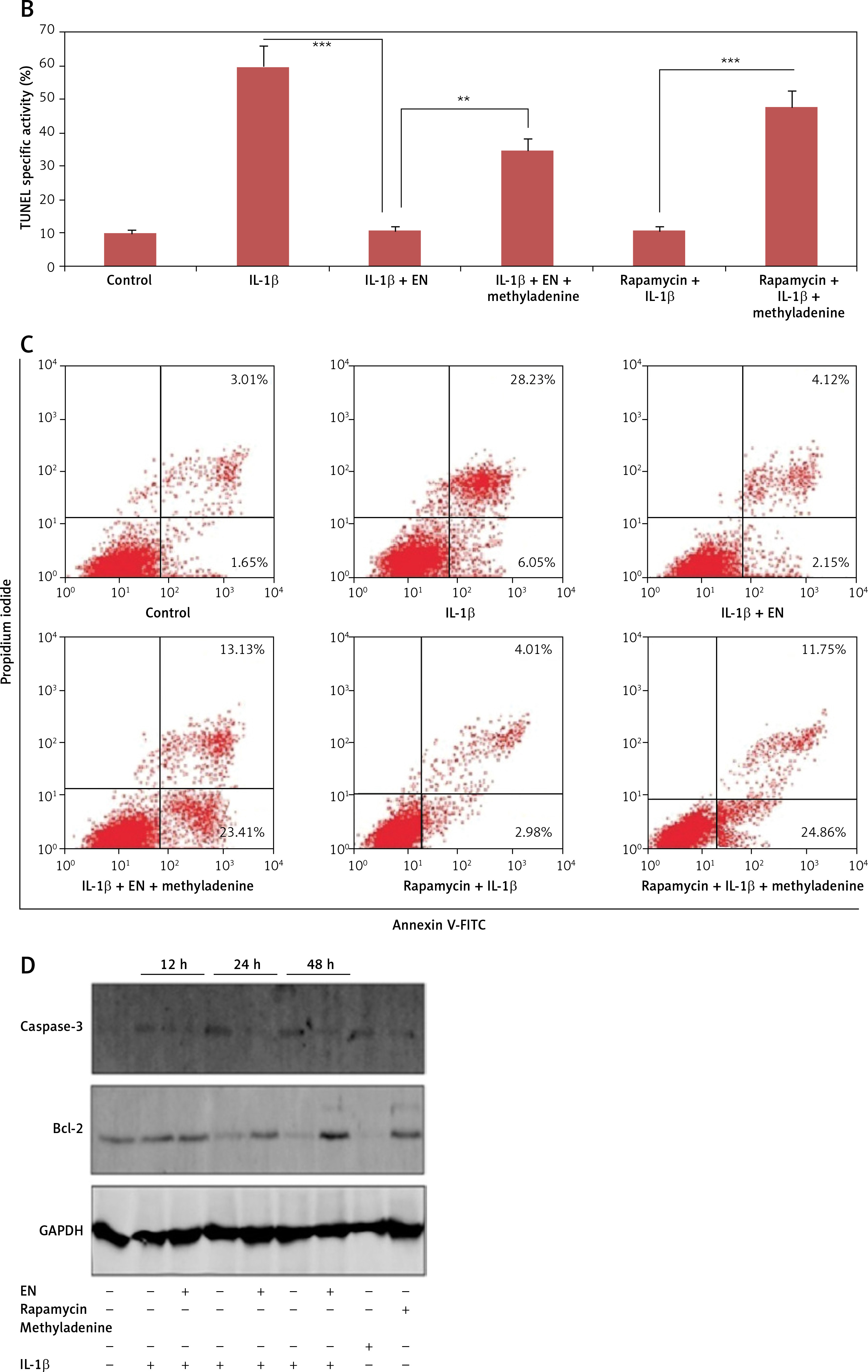
The results of flow cytometry were parallel to the findings of TUNEL assay (Figures 2 B, C). The fluorescence of apoptotic cells labeled with Annexin V and propidium iodide (PI) was estimated by flow cytometry. The study of quadrants suggested that the control cells had the smallest number of cells in lower right (cells with early apoptosis) and upper right (late apoptosis) quadrants, suggesting no signs of apoptosis. The IL-1β treated group showed a high number of cells in lower right and upper right quadrants, suggesting apoptosis. The group receiving treatment with EV along with IL-1β showed a decreased number of cells in both the quadrants, suggesting a protective effect of EN against IL-1β mediated apoptosis. The cells receiving treatment with EN along with IL-1β and 3-methyladenine showed a higher degree of apoptosis, whereas the treatment with rapamycin combined with IL-1β improved the apoptosis, showing a decrease in the number of cells in lower right and upper right quadrants. Furthermore, when the treatment with rapamycin was combined with IL-1β and 3-methyladenine, the number of apoptotic cells increased (Figure 2 C).
Enoxolone suppresses the Bcl-2 mediated apoptotic pathway
To confirm the mechanism by which EN exerts protective action on chondrocytes against apoptosis mediated by IL-1β, the expression of protein Bcl-2 and cleaved caspase-3 was analyzed by western blot after treating the cells with various treatments as summarized (Figure 3 D). The expression of proteins demonstrated that the levels of Bcl-2 declined from 24 h to 48 h in the cells treated with IL-1β (10 ng/ml) against the groups pretreated with EN at the same time intervals (Figures 2 D–F).
Figure 3
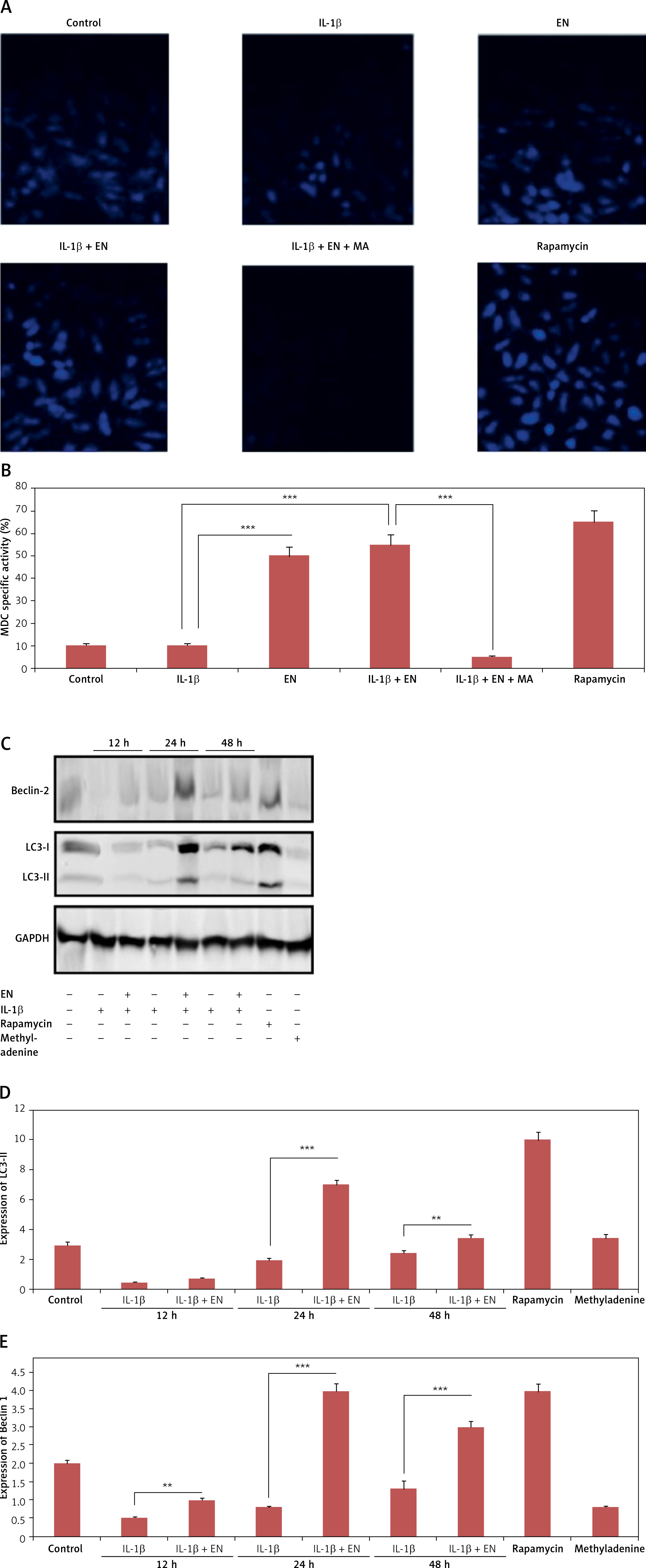
EN resulted in activation of autophagy in chondrocytes. A – Monodansylcadaverine (MDC) staining was done for detection of autophagy induced by EN, B – the results were quantified as % MDC specific activity. C – Expression of Beclin-1 and LC3-I and LC3-II was evaluated after activation of autophagy by western blot analysis at 12, 24 and 48 h under the influence of EN, IL-1β, rapamycin and methyladenine and the levels were quantified (D and E)
Results are presented as means ± SD.*P < 0.05, **p < 0.01, ***p < 0.001.
Enoxolone induces autophagy in isolated chondrocytes
To evaluate whether enoxolone could induce autophagy in isolated Iry chondrocytes, monodansylcadaverine staining, western blot analysis and transmission electron microscopy were performed. It was found that monodansylcadaverine tends to accumulate in the vacuoles of mature autophagocytes and not in the endosome compartments [29]; hence we selected monodansylcadaverine to detect autophagic vacuoles. The results of fluorescence microscopy suggested that the vacuoles of mature autophagocytes stained by monodansylcadaverine appeared as distinct dot-like structures which were spread in the perinuclear regions of cells and also randomly in the cytoplasm. The results demonstrated that (Figures 3 A, B) the treatment with EN caused an increase in the number of monodansylcadaverine stained vesicles after 24 h of treatment, and similar results were observed for cells treated with rapamycin. The outcomes clearly indicated that treatment with EN resulted in induction of vacuoles in mature autophagocytes. We also observed that the treatment with 3-methyl adenine, which is an autophagy inhibitor, blocked the effects of EN. 3-methyl adenine is supposed to inhibit autophagy through its transient inhibiting effect on PI3K, which is essential for initiation of autophagy. The observations by transmission electron microscopy showed presence of apoptosis-induced chondrocytes upon receiving treatment with IL-1β for 24 h, whereas the pre-exposure of cells to EN increased autophagosomes in chondrocytes and decreased the apoptotic bodies. The results of western blot analysis were in agreement with monodansylcadaverine staining; the treatment of cells with EN resulted in a significant increase in levels of beclin-1 and LC3-P (Figures 3 C–E). The results suggested that EN elevated the autophagic activity in chondrocytes.
Enoxolone activates the MAPK/ERK1/2 signaling pathway
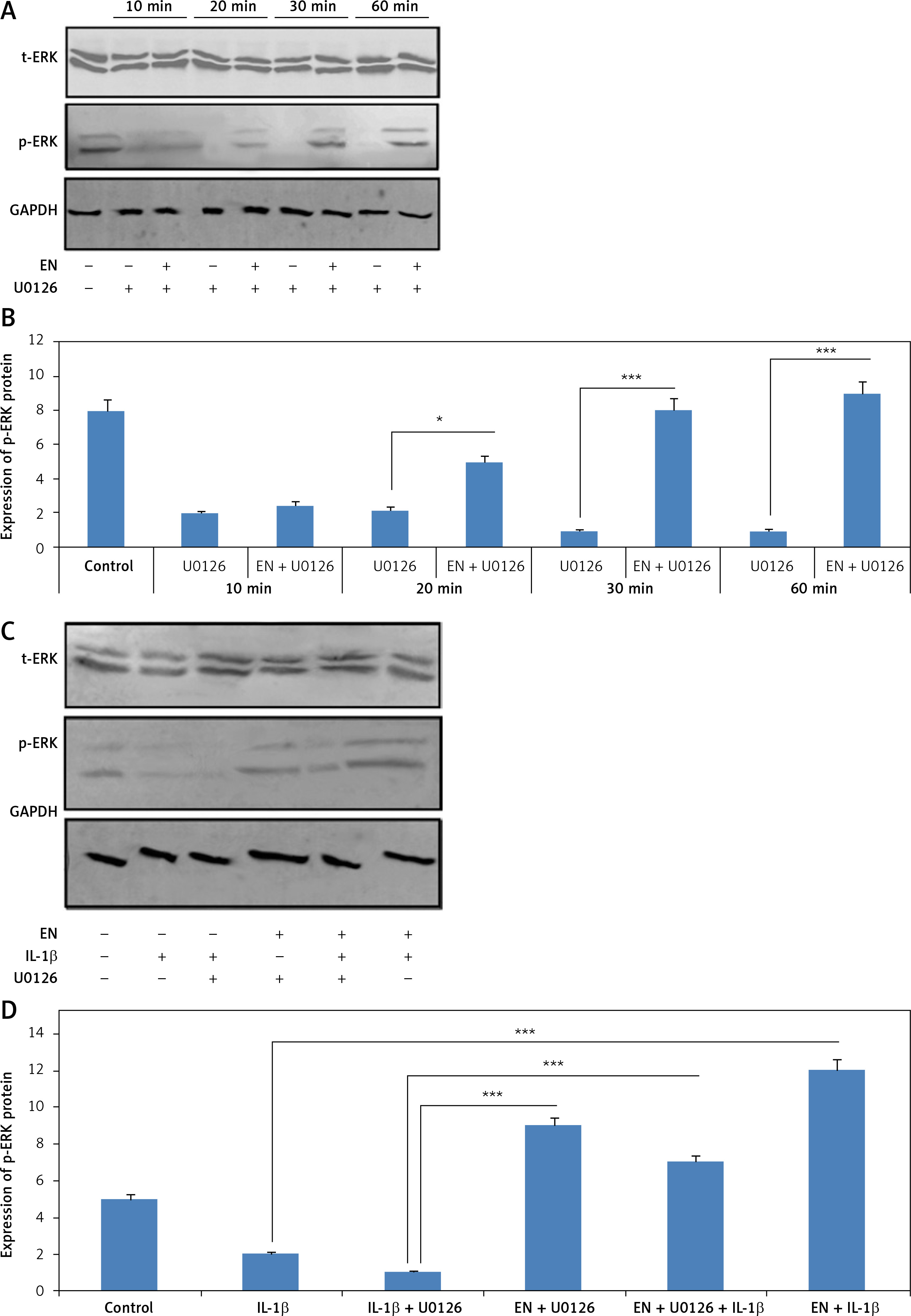
To study the role of EN in the involvement of the MAPK/ERK1/2 pathway, the ERK1/2 inhibitor U0126 was used. It is responsible for suppressing MEK1/2, which further suppresses ERK1/2. For the study, the serum-starved cells were exposed to 10 µM EN and were then incubated with 1 µM of U0126 for 30 min. The results (Figure 4 A, B) suggested that EN stimulated the inhibition of ERK1/2 mediated by U0126 in a time-dependent manner. It is documented that the mitogen-activated protein kinase (MAPK)/ERK1/2 pathway is responsible for regulation of proliferation and cell apoptosis. To study the role of EN in modulating the expression level of ERK1/2, we studied the expression of proteins by western blot, for the same U0126 treated cells with or without exposure to treatment with EN, and examined the expression of ERK1/2 at various time points (Figures 4 A, B). We found that U0126 inhibited the phosphorylation of ERK1/2, whereas the treatment with EN stimulated the phosphorylation of ERK1/2 with increasing time (Figures 4 A, B). We also studied whether the protective action of EN against the treatment with IL-1β was correlated with the phosphorylation of ERK1/2. For the experiment, the Iry chondrocytes were incubated with IL-1β (10 ng/ml) or with U0126 alone or with chondrocytes which received pretreatment with EN (10 µM) for 2 h followed by co-treatment with IL-1β (10 ng/ml) or U0126 for 20, 30 and 60 min. The results suggested that (Figures 4 C, D) pre-treatment with EN unregulated the levels of phosphorylated ERK1/2 significantly in chondrocytes co-treated with IL-1β or stimulated with U0126 against the chondrocytes which received treatment with IL-1β or U0126.
Figure 4
Treatment with EN inverts the inhibitory effect of U0126 in the extracellular signal-regulated kinases 1/2(ERK1/2) in Iry chondrocytes. A – Expression of p-ERK was evaluated by western blot analysis in U0126-exposed chondrocytes. The chondrocytes which were serum starved were pre-exposed and incubated with EN (10 μM) for defined time points followed by co-treatment with 1 μM U0126 for 10 to 60 min. B – Treatment with EN reversed the U0126-mediated inhibition of ERK1/2 in a time-dependent manner, whereas the pan ERK1/2 remained unaltered. C – Effect of treatment with EN on IL-1β mediated inhibition of MAPK/ERK1/2 cascade in Iry chondrocytes in vitro. D – The chondrocytes which were serum-starved and received pre-stimulation of 10 μm EN only for 4 h followed by co-treatment with IL-1β (10 ng/ml) or 10 μM U0126 for 24 h
Results are presented as means ± SD. *P < 0.05, **p < 0.01, ***p < 0.001.
Enoxolone elevates autophagy via activation of the MAPK/ERK1/2 pathway
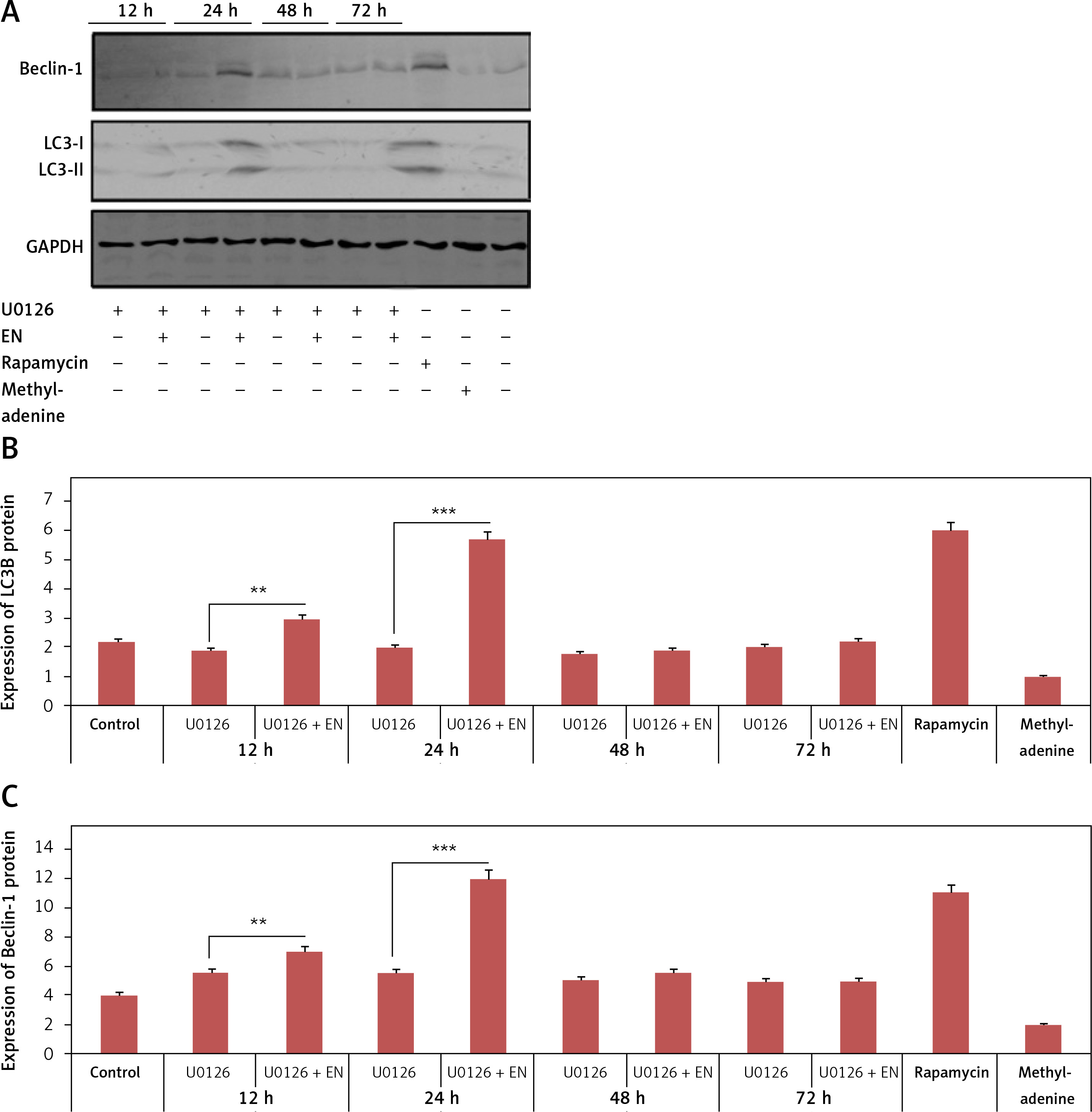
We next postulated that blocking MAPK/ERK1/2 may alter the expression level of LC3-II and Beclin-1, markers of autophagy. Hence the Iry chondrocytes were treated with U0126, alone 12, 24, 48 and 72 h or were given pre-treatment with EN (10 µM). The results suggested that treatment with EN reversed the inhibition of MAPK/ERK1/2 and elevated the expression of LC3-II and Beclin-1 with increasing time; a significant changes in expression was seen at 24 h (Figures 5 A–C), which suggested the involvement and necessity of the MAPK/ERK1/2 pathway for inducing autophagy.
Figure 5
EN could be an activator of autophagy for U0126-mediated inhibition of ERK1/2 in Iry chondrocytes. A – Expression of beclin-1 and LC3-II was analyzed by western blot analysis upon activation of autophagy and the (B and C) levels were quantified
Results are presented as means ± SD. *P < 0.05, * p < 0.01, ***p < 0.001.
Discussion
The study was primarily undertaken to establish the link between enoxolone and autophagy in IL-1β stimulated Iry chondrocytes in vitro. The major outcomes of the study were as follows. (1) The treatment with IL-1β (10 ng/ml) caused morphological changes and resulted in apoptosis of chondrocytes; (2) pre-treatment with enoxolone inhibited the IL-1β mediated apoptosis of chondrocytes with increasing time; (3) enoxolone induced autophagy which resulted in its anti-apoptotic effect; (4) enoxolone inhibited the U0126 mediated down-regulation of p-ERK1/2; (5) the inhibitory effect of IL-1β or U0126 on the phosphorylation of ERK1/2 was reversed by enoxolone.
In the present study we demonstrated that autophagy is essential for inhibiting the apoptosis of chondrocytes. Previously, it has been confirmed that enoxolone possesses an anti-oxidant as well as anti-inflammatory effect [8–10]. The findings of our experiments confirmed that chondrocytes showed autophagy; the findings were in agreement with previous studies which concluded that autophagy is an important feature of chondrocytes [20, 30].
Activation of autophagy is evidenced to play a vital role in maintaining the normal metabolism in chondrocytes. Autophagy acts as a cleaner by removing unwanted damaged proteins and organelles; it plays an important role in preventing disorders such as neurodegeneration, diabetes, autoimmune disorders and cancer. Autophagy is considered to be a well-conserved mechanism and is vital for various processes in the body [31–34]. LC3 and Beclin-1 are two human genes involved in autophagy; both of these genes are prime regulators of the autophagic pathway. The gene beclin-1 combines with type III phosphatidylinositol to form a complex which leads to nucleation of the vesicles. Beclin-1 has a BH3 domain which interacts with Bcl-2 (anti-apoptotic protein), which halts the beclin-1 mediated trigger of autophagy. LC3-I becomes transformed to LC3-II which adheres to the membrane of autophagosomes in the process of activation of autophagy [35].
The articular cartilage has comparatively low turnover of cells and extracellular matrix, which makes it more prone to accumulation of aging-associated changes; due to this fact, the chondrocytes are the only cells present in cartilage whose death is associated with degradation of cartilage [36]. In an earlier report, it was found that the presence of moderate autophagy could inhibit the process of apoptosis in cells [20]. In the present study, we evidenced that a short-term exposure of enoxolone upregulated the autophagic activity in a time-dependent manner in chondrocytes; the upregulation was highest after 24 h, before it declined with time gradually. The outcomes suggested that a short-term exposure to enoxolone could also activate autophagy. Here in the case of chondrocytes, autophagy seems to be a process of self-defense induced by enoxolone against the stimulation by IL-1β. In order to confirm these outcomes, we treated the chondrocytes with rapamycin, which is an activator of autophagy, and 3-methyladenine, which is an inhibitor of autophagy. We observed that, when the cells were pre-treated with enoxolone followed by co-treatment with IL-1β and 3-methyladenine, the apoptosis increased significantly whereas the autophagy was inhibited in the chondrocyte. The results of western blot further confirmed that the levels of caspase-3 in the cells treated with IL-1β were upregulated, whereas the expression levels were suppressed after the cells received treatment with enoxolone. Bcl-2 is a member of the Bcl protein family and is reported to possess anti-apoptotic activity. During the time interval 24 to 48 h, the levels of Bcl-2 decreased in cells exposed to IL-1β, whereas the pre-exposure of enoxolone reversed the expression of Bcl-2. In a report earlier, cytochrome c was found to participate in the activation of caspase-3, whereas Bcl-2 was reported to inhibit the release of cytochrome-c from mitochondria [37].
The mitogen-activated protein kinase (MAPK) pathway is vital for survival and differentiation of chondrocytes. Inhibition of the MAPK pathway causes apoptosis of human chondrocytes [38]. The MAPK/ERK pathway can block apoptosis in many cell types [39]. In the present study, we evaluated the potential role of ERK in autophagy of chondrocytes by treating them with U0126, which is a highly selective inhibitor of MAPK. We observed that treatment with U0126 showed a decrease in markers of autophagy compared to the chondrocytes receiving pre-treatment with enoxolone, which indicated existence of a functional death pathway associated with autophagy of chondrocytes. A previous study established that blocking the ERK1/2 cascade could bring about apoptosis of chondrocytes [38, 39]; the findings were in agreement with the outcomes of our experiments, where we proved that autophagy and apoptosis have an inverse relationship [40].
Altogether, the outcomes of our study established the importance of autophagy in the survival of chondrocytes; also inhibition of autophagy leads to caspase-dependent death. The results also suggested that self-activation of autophagy acts as a protective mechanism against apoptosis when treated with enoxolone for 48 h (short duration). These outcomes suggest that the anti-apoptotic effect of enoxolone is channeled through the MAPK/ERK1/2 pathway.
In conclusion, the present study revealed that the treatment with enoxolone elevated autophagy in the chondrocytes, and also, autophagy acted as a pro-survival path in chondrocytes induced by IL-1β. The study also confirmed that enoxolone enhanced autophagy and protected the chondrocytes against IL-1β mediated apoptosis through ERK1/2 mediated autophagy. Hence we propose that increasing autophagy via increasing the expression of p-ERK1/2 could be an important strategy in treating osteoarthritis. Though our ongoing research will clarify the biological actions of enoxolone, a clinical study is necessary to confirm our current in vitro findings.