Current issue
Archive
Manuscripts accepted
About the Journal
Editorial office
Editorial board
Section Editors
Abstracting and indexing
Subscription
Contact
Ethical standards and procedures
Most read articles
Instructions for authors
Article Processing Charge (APC)
Regulations of paying article processing charge (APC)
BIOLOGY MOLECULAR / RESEARCH PAPER
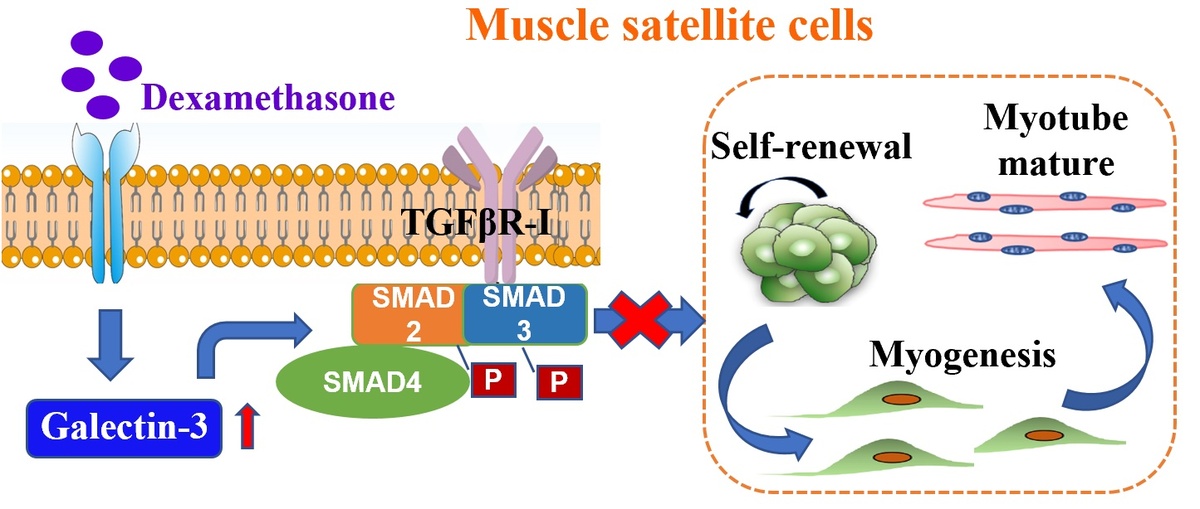
Galectin-3 regulates skeletal muscle loss via TGFβ1-Smad2/3 signaling activation in mice
1
Shanghai Key Laboratory of Clinical Geriatric Medicine, Department of Geriatric Medicine, Huadong Hospital, Fudan University, China
2
Institute of molecular medicine and innovative pharmaceutics, Qingdao University, China
3
Department of Endocrinology, Huadong Hospital, Fudan University, China
4
National Clinical Research Center for Ageing and Medicine (Huashan), China
Submission date: 2022-10-20
Final revision date: 2023-02-07
Acceptance date: 2023-02-24
Online publication date: 2023-03-26
Corresponding author
Zhijun Bao
Shanghai Key Laboratory of Clinical Geriatric Medicine, Department of Geriatric Medicine, Huadong Hospital, Fudan University, Shanghai, China
Shanghai Key Laboratory of Clinical Geriatric Medicine, Department of Geriatric Medicine, Huadong Hospital, Fudan University, Shanghai, China
KEYWORDS
TOPICS
ABSTRACT
Introduction:
Endogenous glucocorticoids (GCs) played a pivotal role in the pathogenesis of skeletal muscle loss. However, to date, the underlying molecular mechanisms underlying are not yet fully understood. Galectin-3 (Gal-3) is a member of a beta-galactoside-binding animal lectins, consistently associated with inflammation and fibrosis in the pathogenesis of various disease states. The present study aimed to explore the role of Gal-3 in GC-induced skeletal muscle loss.
Material and methods:
Myogenic differentiation capacity was detected after in vitro Gal-3 knockdown (KD) or in vivo administration of Gal-3 inhibitor. The activation of transforming growth factor type beta 1 (TGFβ1) and Smad2/3 signaling pathways was determined by western blot, co-immunoprecipitation, and immunofluorescence.
Results:
Gal-3 was up-regulated during dexamethasone (Dex) administration in mice. In the established GC-induced muscle loss model, Gal-3 inhibition recovered grip strength and muscle mass. In vitro, Gal-3 KD promoted the myogenic differentiation capacity of C2C12 myoblasts, and prevented the reduction of fully differentiated myotubes. Gal-3 results in overexpression of TGFβ1 and TGFβR-I, which affects Smad2, 3 phosphorylation and subsequently mediates skeletal muscle reduction by activating the Smad2/3 signaling pathways.
Conclusions:
The present study demonstrated that Dex elevated Gal-3 levels in skeletal muscle. Gal-3 facilitates the activation of TGFβ1-Smad2/3 signaling pathways in myoblasts, and contributes to myogenesis inhibition and skeletal muscle loss. This study raises awareness about the follow-up of patients receiving GC therapy. Further, inhibition of Gal-3 provides a possible therapeutic strategy for skeletal muscle loss.
Endogenous glucocorticoids (GCs) played a pivotal role in the pathogenesis of skeletal muscle loss. However, to date, the underlying molecular mechanisms underlying are not yet fully understood. Galectin-3 (Gal-3) is a member of a beta-galactoside-binding animal lectins, consistently associated with inflammation and fibrosis in the pathogenesis of various disease states. The present study aimed to explore the role of Gal-3 in GC-induced skeletal muscle loss.
Material and methods:
Myogenic differentiation capacity was detected after in vitro Gal-3 knockdown (KD) or in vivo administration of Gal-3 inhibitor. The activation of transforming growth factor type beta 1 (TGFβ1) and Smad2/3 signaling pathways was determined by western blot, co-immunoprecipitation, and immunofluorescence.
Results:
Gal-3 was up-regulated during dexamethasone (Dex) administration in mice. In the established GC-induced muscle loss model, Gal-3 inhibition recovered grip strength and muscle mass. In vitro, Gal-3 KD promoted the myogenic differentiation capacity of C2C12 myoblasts, and prevented the reduction of fully differentiated myotubes. Gal-3 results in overexpression of TGFβ1 and TGFβR-I, which affects Smad2, 3 phosphorylation and subsequently mediates skeletal muscle reduction by activating the Smad2/3 signaling pathways.
Conclusions:
The present study demonstrated that Dex elevated Gal-3 levels in skeletal muscle. Gal-3 facilitates the activation of TGFβ1-Smad2/3 signaling pathways in myoblasts, and contributes to myogenesis inhibition and skeletal muscle loss. This study raises awareness about the follow-up of patients receiving GC therapy. Further, inhibition of Gal-3 provides a possible therapeutic strategy for skeletal muscle loss.
Share
RELATED ARTICLE
| eISSN: | 1896-9151 |
| ISSN: | 1734-1922 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.