Introduction
Hypopituitarism in neonates is rare. It affects between 1 in 4000 to 1 in 10,000 live births [1] and can manifest as a partial or complete insufficiency of pituitary hormone secretion. It is, however, a serious condition with life-threatening complications if untreated. Although 52% of neonates with hypopituitarism present postnatally with associated complications, only 23% are diagnosed in the neonatal period [2]. Neonates are often asymptomatic or present with non-specific symptoms. This review focuses on the genetic causes of hypopituitarism including those causing isolated pituitary problems and those with extra-pituitary manifestations and assesses the evidence base to determine which infants in whom a genetic cause should be suspected and investigated.
Presentation
Congenital hypopituitarism may present with a loss of function of a single pituitary hormone, commonly isolated growth hormone deficiency (IGHD), or with two or more pituitary hormone deficiencies (multiple pituitary hormone deficiency – MPHD). It may be part of an underlying syndrome with abnormalities of extra-pituitary structures such as the eyes and forebrain which have a common embryological origin to the pituitary gland. Neonates may often be asymptomatic and then develop abnormal signs due to evolving hormone deficiencies. Congenital hypopituitarism may present at birth, but is difficult to diagnose in the neonatal period, as the signs may be non-specific, including lethargy, apnoea, poor weight gain and feeding problems and the clinical presentation depends on the pituitary hormone deficiencies present.
Growth failure due to GHD usually presents after the first year after birth, though may be present in infancy in severe GHD [3]. Severe hypoglycaemia, at or soon after birth, is a uncommon presentation and results from adrenocorticotropic (ACTH) deficiency or lack of growth hormone (GH) secretion [4]. Micropenis, defined as a stretched penis length of less than 2.5 cm, may be present and is caused by low luteinizing hormone (LH) secretion which is responsible for maintaining fetal testosterone production. Bilateral undescended testis may also be present as the fetal testicular descent is dependent on LH. Less specific signs of hypopituitarism include neonatal hepatitis and cholestatic jaundice [5]. In one study, jaundice was found to be the most common presentation in 35% of those presenting with hypopituitarism as neonates or young infants [6]. The pathophysiology of liver damage in hypopituitarism is suggested to be due to insufficient glucocorticoid and thyroid hormones [7]. In patients with thyroid stimulating hormone (TSH) deficiency, hyperbilirubinaemia can be associated with temperature instability [8]. The most common electrolyte imbalance is hyponatraemia, which is related to inadequate levels of ACTH and TSH. In those with posterior pituitary abnormalities, diabetes insipidus is often the key presentation, due to lack of antidiuretic hormone (ADH).
Genetic abnormalities
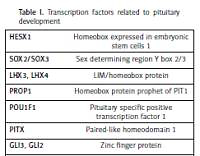
Mutations in any of the genes involved in pituitary development may result in congenital hypopituitarism leading to deficiency in one or more pituitary hormones. Genetic abnormalities, however, are only identified in 5–10% of those with congenital hypopituitarism, but it is estimated that a further 5–30% of individuals with a first-degree relative with the disorder possess a genetic mutation yet to be identified [8]. Mutations in gene coding for growth hormone (GH1) and gene coding the growth hormone releasing hormone receptor (GHRHR) are known to cause idiopathic growth hormone deficiency (IGHD). Mutations in various genes which encode transcription factors related to pituitary development have been associated with varying degrees of hypopituitarism (Table I). In one study, 13% of individuals had a mutation in one of five commonly mutated genes (POU1F1, PROP1, LHX3, LHX4, and HESX1), rising to 52% of individuals in familial cases of multiple pituitary hormone deficiency (MPHD) [9]. The prevalence of genetic mutations varies worldwide, such that PROP1 mutations account for the majority of genetic MPHD in Europe, but are much less common in Asians [10]. We have focused on the role of the main transcription factors involved in pituitary development and mechanisms underlying hypopituitarism and accompanying phenotypes.
Table I
Transcription factors related to pituitary development
PROP1
PROP1, located on chromosome 5q, encodes for a paired-like homeodomain protein essential for HESX1 repression and POU1F1 expression [10, 11]. It is essential for the development of most types of anterior pituitary cells, therefore, patients can present with deficiency of all hormones secreted by the anterior pituitary [3]. In humans, at least 25 mutations in PROP1 (both homo- and heterozygous) have been described and these are responsible for the majority of MPHDs [12]. The most frequent mutations are a two-base-pair deletion within exon 2, causing a frameshift in codon 101 (301-302delAG) and a one-base-pair deletion in codon 50 (150delA). Both result in the production of a termination codon and early cessation of protein sequencing [10, 11]. Mis-, nonsense and splicing error mutations have also been found [10]. Recessive PROP1 mutations often present with GH deficiency early in life and may have the phenotypic presentation of gonadotrophin deficiency (micropenis, cryptorchidism, lack of puberty and/or infertility). TSH and ACTH are often normal in early life, but deficiencies in these hormones often evolve with increasing age emphasising the need for continuous monitoring.
POU1F1
POU1F1 is another pituitary-specific transcription factor, responsible for differentiation of somato-lactotrophs and thyrotophs. These cells secrete GH, PRL and TSH. POU1F1 may also be involved in autoregulation of expression, as its binding sites have been found on the POU1F1 gene. Approximately 8% of MPHD and isolated GH cases are due to mutations in this gene [13]. Since first being described, 38 mutations have been reported; most are autosomal recessive, although five dominant mutations have been found [11, 14, 15]. A sibling case suggests that isolated mutations in the short isoform of POU1F1 might be sufficient for induction of POU1F1 related combined pituitary hormone deficiency [16]. As in PROP1 mutations, individuals may present with GH deficiency including early growth failure and failure to thrive. The age at presentation of TSH deficiency is highly variable [14, 17] and cases with normal TSH levels have been reported [13]. Indeed, the pituitary gland may be normal or hypoplastic on MRI [11, 13].
LHX3 and LHX4
LHX3 and LHX4 are involved in the complex genetic cascade resulting in pituitary development, critical for development of Rathke’s pouch. LHX4 is essential for pituitary development and motor neuron specification in mammals [18]. Extra-pituitary expression has been documented in other regions of the brain and spinal cord. Mutations in genes encoding for these transcription factors are rare. Seven different mutations in LHX3, including base pair deletions, mutations and even one case of whole gene deletion have been documented [19]. Mutations in LHX3 present similarly to mutations in PROP1 with GH, PRL, TSH and/or LH/FSH deficiency. Rarely, ACTH deficiency has also been described. In addition to pituitary abnormalities, patients present with a short, rigid cervical spine, limited head rotation and trunk movement [14, 19]. Bilateral sensorineural hearing loss has also been documented in some cases [19]. Other abnormalities include cervical vertebral malformations (rigid neck, scoliosis), mild developmental delay and moderate sensorineural hearing loss [20]. Heterozygous LHX3 mutations may lead to a mild phenotype of combined pituitary hormone deficiency [21]. Sonic hedgehog (SHH) is an essential morphogenetic signal which dictates cell fate decision in several organs in mammals. In vitro data suggest that SHH is required to specify LHX3+/LHX4+ Rathe’s Pouch (RP) progenitor identity. In mouse embryos, conditional deletion of SHH in the anterior hypothalamus results in a fully penetrant phenotype characterised by complete arrest of RP development with lack of LHX3/LHX4 expression and total loss of pituitary development [22]. In one case of LHX4 mutation, a fully penetrant, dominant mutation caused defective splicing. An magnetic resonance imaging (MRI) showed anterior pituitary hypoplasia, an ectopic posterior pituitary and a shortened pituitary stalk. The three-generation family all presented with short stature, some with an abnormal sella turcica and Chiari malformation with pointed cerebellar tonsils [23]. In another case, who presented with severe respiratory distress and hypoglycaemia soon after birth, had multiple pituitary hormone deficiencies. MRI demonstrated a hypoplastic anterior pituitary, an ectopic posterior lobe, a poorly developed sella turcica and Chiari malformation. Sequence analyses of the LHX4 gene identified a heterozygous missense mutation (P366T) in exon 6, which was present in the LIM4 specific domain [18]. Screening of 136 patients with congenital hypopituitarism associated with malformations of brain structures, pituitary stalk or posterior pituitary gland demonstrated a novel dysfunctional LHX4 mutation (p.Thr99fs) which was responsible for the patient’s phenotype [24]. In another study, heterozygous missense mutations in LHX4 resulting in the substitution of amino acids resulted in GH deficiency, +/– reductions in LH, TSH, FSH or ACTH and aberrant pituitary morphology [25]. Screening of 417 unrelated patients with isolated growth hormone deficiency or combined protein deficiency associated with ectopic posterior pituitary and/or sella turcica anomalies demonstrated seven heterozygous variations in LHX4 [26].
Homeobox expressed in ES cells 1 (HESX1) and sry-type HMG box (SOX) genes in septo-optic dysplasia
Septo-optic dysplasia, although rare, is the most common congenital cause of hypopituitarism, encompassing any combination of two of the triad: optic nerve hypoplasia, hypopituitarism and midline structural abnormalities [1]. Its incidence is reported to be 1 in 10,000 live births. Hypopituitarism affects 62% of those with the condition and its severity may be predicted by the degree of midline structural abnormalities. Several genetic mutations have been demonstrated to result in various forms of the condition [27].
HESX1, a transcriptional repressor, regulates some of the earliest stages of pituitary development. It is expressed during gastrulation in the region which develops into the forebrain, after which it is restricted to expression in the ventral diencephalon and the region of the oral ectoderm which forms Rathke’s pouch [14]. It has a role in optic nerve formation [3, 14]. Null HESX1 mice have pituitary hypoplasia, micro- or anophthalmia and agenesis of midline forebrain derivatives; a similar phenotype to that of the human condition septo-optic dysplasia [14, 28]. A large study confirmed that the overall expression of HESX1 mutations in SOD is low [9, 29]. Another study looked at the prevalence of pituitary hormone abnormalities with optic nerve hypoplasia (with or without midline brain abnormalities), however, found that the prevalence may be as high as 72% [30]. Several different mutations have been documented; which vary in the extent of hormone deficiency [14]. Isolated GH deficiency to MPHD have been described with or without optic nerve hypoplasia and or mid-line brain abnormalities [14, 31]. Heterozygous mutations are milder than the bi-allelic type.
SOX2 and -3 are SRY-related HMG box transcription factors may be responsible for maintaining pituitary progenitor cells and their function [12]. Heterozygous mutations in SOX2 genes are associated with developmental eye abnormalities such as anophthalmia and microphthalmia. Oesophageal abnormalities, urogenital defects, developmental delay and sensorineural hearing loss have also been associated with SOX2 mutations, as SOX2 plays an important role in the development of the eye, forebrain, gastrointestinal tract and urogenital systems. The phenotype usually includes anterior pituitary hypoplasia on MRI scan and hypogonadotrophic hypogonadism. GHD has been described in a single case [32]. SOX3 is located on the X-chromosome, therefore, mutations in SOX3 primarily affect males. Under and over exposure of the gene has been involved in causing X-linked hypopituitarism, resulting in varying degrees of hypopituitarism, mid-line brain abnormalities and learning disabilities
SHH and GLI2 in holoprosencephaly (HPE)
HPE encompasses a number of structural midline forebrain abnormalities, some of which are fatal in utero. The most severe form is alobar HPE, whereby the forebrain fails to develop into two hemispheres. This is often accompanied by midline facial deformities. The milder forms, which are compatible with life, can present with defective facial features such as hypotelorism and one nostril, but can also present with more distal manifestations such as polydactyly. Due to the location of the hypothalamus and pituitary gland as midline structures, their formation may be affected in HPE, resulting in hypopituitarism. Deficient ADH production may occur in as many as 70% of affected patients [33]. Other variations of hypopituitarism presentation are isolated GH deficiency, MPHD or increasing manifestation of hormone deficiencies with age. Genetic mutations in the sonic hedgehog (SHH) gene are known to cause HPE and have also been linked to pituitary gland formation [34]. SHH is expressed in midline structures early in development, as well as controlling distal limb development. It is expressed in the ventral diencephalon and oral ectoderm, prior to formation of Rathke’s pouch. SHH signalling, together with FGF8/10 (also known to cause HPE when mutated), regulate LHX3 signalling.
The GLI2 transcription factor, along with GLI1 and GLI3, is essential for mediating SHH signalling [35–37]. In mice, whilst GLI1 and GLI3 are unessential, GLI2 has been shown to be the major transcription factor required for pituitary development [38]. GLI2 is expressed in the same locations as SHH during pituitary development. Mutations in this gene phenotypically present with HPE-like features: polydactyly and hypopituitarism [35, 39]. GLI2 mutations, although often found in GHD with normal brain morphology [40], can cause anterior pituitary hypoplasia and an absent posterior pituitary. GLI2 mutants have been shown to affect the proliferation and specialization of cells in the pituitary gland, but not affect its formation. Therefore, the absence of the posterior pituitary is hypothesized to be due to its relationship with SHH [41].
GLI3 and Pallister-Hall syndrome
GLI3 autosomal dominant mutations causes Pallister-Hall syndrome [42]: a rare condition characterized by polydactyly, bifid epiglottis, hypothalamic hamartoma, pituitary dysfunction and an imperforate anus. The endocrine abnormalities can range from isolated GH deficiency to MPHD. De novo mutations are more severe in presentation than inherited mutations [43].
FGF8 and FGFR1 in Kallmann syndrome and idiopathic hypogonadotrophic hypogonadism (IHH)
Kallmann syndrome is one of a group of conditions known as hypogonadotropic hypogonadism (HH), whereby gonadal development and functional activity is impaired by reduced gonadotropin secretion, associated with reduced or lack of sense of smell. Mutations in FGF8 and FGFR1 have been found in 10% of patients with Kallmann syndrome and 7% of those with idiopathic hypogonadotropic hypogonadism [44]. They are believed to control release of gonadotropin releasing hormone, which stimulates LH and FSH release from the pituitary gland. Clinically, HH presents with failure of pubertal onset with low levels of sex steroids and gonadotrophins. Milder phenotypes may include secondary amenorrhea, delay of puberty onset or adult onset of hypogonadotrophic hypogonadism. Although previously mutations in these genes were only thought to cause Kallmann syndrome/ IHH, more recent studies have shown that mutations in these genes can also cause a wider phenotype of MPHD or SOD [45]. Mutations in several other genes are also impacted in the pathogenesis of Kallmann syndrome and IHH [46].
Investigation of suspected hypopituitarism
In a neonate with suspected hypopituitarism, biochemical evaluation of the hypothalamic-pituitary axis should be undertaken to evaluate for and define the extent of pituitary hormone deficiencies. As cortisol deficiency can be life-threatening, evaluation of ACTH deficiency by a short Synacthen test (sensitivity of 80%) is essential. TSH deficiency can be identified by low serum free thyroxine (fT4) and normal/ low serum TSH. GH stimulation tests to diagnose GHD are undertaken after a year of age, although low GH levels during earlier spontaneous hypoglycaemia may suggest GHD. Hypogonadotrophic hypogonadism may be suspected in males with micropenis with or without bilateral undescended testes. Baseline low gonadotrophins with GnRH and HCG (in males) stimulation tests in infancy can help determine gonadotrophin deficiency. Diabetes insipidus is diagnosed by the presence of hypernatremia with raised plasma osmolality and concurrent low urine osmolality. MRI imaging of the brain and pituitary gland should be undertaken in any neonate with suspected hypopituitarism to evaluate the size of the anterior pituitary gland, the presence and location of posterior pituitary gland, the presence of midline (corpus callosum/ septum pellucidum) and other brain (such as HPE) abnormalities and the presence and size of the optic nerves.
Although several genetic causes of hypopituitarism have been identified by novel genetic technologies, the yield of genetic testing in hypopituitarism remains low. Detailed clinical and biochemical phenotyping along with MRI imaging of the pituitary gland may help identify those where genetic testing may be helpful. For example. PROP1 mutation needs to be considered in a patient with multiple pituitary hormone deficiency with an intact pituitary stalk and posterior pituitary [47]. In patients with septo-optic dysplasia, a HESX1 mutation should be investigated, although it has been recently shown that HESX1 mutations cause variable clinical features in patients and can also be seen in patients without septo-optic dysplasia [48]. Genetic testing is also likely to be beneficial in familial cases of hypopituitarism. Genetic analysis, however, is unlikely to be helpful in sporadic cases of hypopituitarism in Western European populations in the absence of specific MRI and extra-pituitary abnormalities [49, 50].
Management of an infant identified with hypopituitarism requires a multi-disciplinary team approach in a tertiary paediatric endocrinology centre and includes adequate replacement of hormone deficiencies and management of associated conditions and/or neurodevelopmental delay. Life-long monitoring for adequacy of hormone replacement and evaluation of evolving hormone deficiencies is indicated. Genetic counselling should be offered to parents in whose infant a genetic cause has been identified.
Conclusions
Due to the serious complications associated with untreated disease, hypopituitarism should be considered in neonates presenting with hypoglycaemia, hypogonadism or unexplained jaundice. Many factors can cause congenital hypopituitarism: although rare, genetic causes of hypopituitarism are becoming better understood. The syndromes presenting with hypopituitarism associated with extra-pituitary abnormalities are more easily characterized and genetic causes more likely. For PROP1 it is important to consider the geographical origin of the patient as this mutation is more frequent in some ethnic groups [49]. MRI imaging of the brain and pituitary gland, along with hormonal profile and presence of specific extra-pituitary manifestations can help target specific genetic testing in patients with hypopituitarism. Similarly, this should be undertaken in infants with suspected hypopituitarism due to a positive family history. Despite the identification of an increased number of genetic causes of isolated or multiple pituitary deficiencies it should be appreciated that the etiology of most (80–90%) congenital causes of hypopituitarism remain unsolved [12].