Introduction
Marine ecosystems occupy a significant portion, around 71%, of the Earth’s surface. These diverse habitats are sustained by the vital functions performed by various bacterial populations [1]. Exopolysaccharide (EPS), a principal organic compound produced by ocean microorganisms, accounts for approximately half of the primary generation of organic matter. These polymers play a crucial role in maintaining marine environments by facilitating sedimentation, particle formation, and the cycling of dissolved metals and organic carbon [2]. Exopolysaccharides are essential for the growth and survival of organisms in harsh marine environments. These polymers facilitate crucial functions such as nutrient uptake, aggregation, adherence to surfaces, and the production of biofilms, which are vital for the survival and thriving of marine organisms [3].
Microbial polysaccharides are hydrophilic biopolymers that may be intracellular, structural, or exopolysaccharides, exhibiting various intriguing properties, such as biocompatibility, biodegradability, and nontoxicity [4]. When comparing EPS with the first two groups, EPS exhibits a broader range of applications and employs more extensive methodologies for extraction and processing [5–7]. Numerous fungi, algae, and Gram-positive and Gram-negative bacteria can produce EPS [8]. The formidable, harsh marine ecosystem has the potential to elicit the synthesis of EPSs by marine microorganisms [3]. They provide microorganisms the potential to enhance their tolerance towards biotic and abiotic stressors [9]. Most microbial EPSs exhibit heterogeneity in their composition, consisting of diverse monosaccharides such as glucose, galactose, glucuronic acid, and others, arranged in a specific and characteristic ratio [10].
Microbial EPSs typically have high molecular weights, ranging from 10 to 6,000 kDa [11]. The anionic nature of most reported EPS is primarily attributed to the presence of pyruvate and uronic acid moieties linked to ketals and the inclusion of inorganic residues such as sulfate or phosphate groups [12]. Due to the growing demand for natural polymers in industries such as food and pharmaceuticals, there has been a recent surge in interest in microbially produced polysaccharides [12].
The recovered EPS from various bacterial strains exhibited substantial physicochemical and structural variations [13]. Marine EPSs show a considerably higher degree of complexity and diversity in bioactivities compared to terrestrial origins [14]. These bacterial metabolites may have potential uses as anti-inflammatory, antioxidant, antimicrobial, and anticytotoxic agents, in addition to various other pharmacological applications. The most well-known producers of EPSs are bacteria belonging to the genera Lactobacillus, Bifidobacterium, Leuconostoc, Pediococcus, Streptococcus, Enterococcus, and Weissella [15, 16]. Furthermore, EPS generated by specific Lactobacillus species, such as Lactobacillus acidophilus, Lactobacillus gasseri, Lactobacillus plantarum, and Lactobacillus rhamnosus, isolated from diverse sources, has been shown to exhibit antitumor as well as antioxidant properties [17]. Interestingly, EPSs produced by Lactobacillus plantarum, Lactobacillus acidophilus, and Lactobacillus helveticus are the most commonly reported EPSs with good anticancer properties among EPS-producing species [18]. Even within the same bacterial species, the antiproliferative activity of the EPS can vary from strain to strain [19].
Building upon this foundation, existing research has revealed that microbial EPSs possess a diverse range of therapeutic potential, including antibacterial [20, 21], antioxidant [22, 23], anti-inflammatory [24, 25], anticancer [23, 24], and gel-forming attributes [26]. Furthermore, numerous studies have documented the ability of certain EPSs to modulate wound cellular metabolism, facilitating tissue repair and regeneration and accelerating the healing process [27, 28]. In addition, recent investigations have successfully isolated sulfated polysaccharides exhibiting notable anticoagulant functionalities from various marine species [29–31]. These investigations aimed to identify an alternative anticoagulant to heparin, a glycosaminoglycan (GAG) family member characterized by its sulfated polysaccharide structure [32]. Several factors make heparin alternatives preferable. Some religious groups avoid heparin because it comes from pig intestines and bovine lungs. Additionally, heparin is linked to fatal disorders. Liu et al. found that critical COVID-19 patients treated with heparin have a high mortality risk from thrombocytopenia [33]. These drawbacks have spurred researchers to find safer, more effective alternatives [30, 34, 35].
Given the remarkable biomedical applications of microbial EPSs and the perpetual efforts to discover and explore new bioactive bacterial EPSs, the current investigation aimed to isolate novel bioactive compounds from marine bacteria collected from the Red Sea, with the potential for development into pharmaceutical and therapeutic drugs. The specific objectives of this study were as follows: primary screening and isolation of the marine bacterium with the highest EPS production, followed by 16S rRNA molecular identification; chemical characterization and analysis of the generated EPS using Fourier-transform infrared (FT-IR) spectroscopy, high-performance liquid chromatography (HPLC); assessment of the EPS’s antioxidant properties through 2,2-diphenyl-1-picrylhydrazyl (DPPH), total antioxidant capacity (TAC), and ferric reducing antioxidant power (FRAP) assays, as well as its anti-inflammatory potential using human red blood cell (HRBC) hemolytic and membrane stabilization assays; evaluation of the EPS’s anticoagulant activity through classical prothrombin time (PT) and partial thromboplastin time (PTT) assays, and investigation of its wound healing potential; in vitro assessment of the EPS’s lipase inhibitory activity, as well as its antidiabetic effects through α-amylase and α-glucosidase inhibition studies; and examination of the EPS’s antimicrobial and antibiofilm properties against a panel of Gram-positive (G+ve) and Gram-negative (G-ve) pathogenic bacteria, including Helicobacter pylori.
Material and methods
Sampling of Red Sea bacteria and selection of isolates for molecular analysis
Bacterial specimens were obtained and separated from sand samples from the Red Sea using the serial dilution technique [36]. The bacterial strains were carefully chosen considering their culture growth characteristics and their maximum production rate of EPS.
The bacterial genetic classification was conducted by employing a 16S rRNA sequence, which was subsequently subjected to additional phylogenetic analysis [37]. Using the BLAST tool, the acquired DNA sequence was compared with the GenBank database at the NCBI. Subsequently, sequence alignment was conducted to assess the degree of similarity between the isolate’s sequence and those present in the database.
Production, extraction, and physicochemical characterization of bacterial EPS
For producing EPS, the promising strain R9 was selected. The final step involved the addition of the fermentation medium broth, as described by Liu et al. [38]. A total of 4 liters of ethanol was introduced into the supernatant for fractional precipitation. The UV absorption spectra in the 200 to 800 nm wavelength range were examined to determine the presence of proteins and nucleic acids [39]. FTIR spectra were analyzed utilizing the FTIR-UNIT Bruker Vector 22 Spectrophotometer [40]. The identification of uronic acid in the EPS was achieved by employing the colorimetric method described by Filisetti-Cozzi and Carpita [41]. The sulfate content was quantified using Garrido’s method [42]. The methodology described by Randall et al. was employed to investigate the monosaccharide content of the specimen using an Aminex carbohydrate HP-87C column (300 × 7.8 mm) at a flow rate of 0.5 ml/min. Water was employed as the eluent, and the detector was a refractive index (RI) detector. Acid hydrolysis was performed by hydrolyzing a known quantity of EPSR9 (15 mg) with HCOOH (88%) in a sealed vessel at 100°C for 5 h. Afterward, the hydrolysate was quantitatively transferred to a crucible and HCOOH evaporated to dryness under vacuum at 40°C. The hydrolysate was then washed with dH2O and concentrated under vacuum after repeatedly evaporating to eliminate the formic acid. The sample was frozen in a sealed vial for later analysis. Next, HPLC was used to separate and quantify the EPSR9 hydrolysate by analyzing the mono sugars on an Agilent Pack series 1200 instrument equipped with an Aminex carbohydrate HP-87C column (300 mm × 7.8 mm). Peaks were identified by comparing retention times to known reference standards. Concentrations of sugars were calculated from retention times and peak areas using Agilent Packard data analysis [43].
Antioxidant evaluation of the EPS
DPPH test
The antioxidative capacity of the EPS was assessed at various concentrations (1.95–1000 µg/ml) using the methodology described by Brand-Williams [44]. The spectrophotometer (UV-VIS Milton Roy) was employed to measure the absorbance at a wavelength of 517 nm. The experimental procedure involved applying ascorbic acid as the reference standard, and the testing method was carried out in triplicate. The IC50 value of the EPS was determined by constructing a logarithmic dose-inhibition curve.
TAC examination
The quantitative examination of the EPS was performed using spectrophotometric analysis adopting the phosphomolybdenum approach described by Prieto et al. [45]. The quantification of absorbance at a wavelength of 630 nm was performed using a microtiter plate reader (Biotek ELX800; Biotek, Winooski, VT, USA). Calculation of the values was performed using the ascorbic acid equivalent (AAE) unit, expressed in µg/mg of the tested EPS, as outlined by Lahmass et al. [46].
FRAP assay
To investigate the impact of solvent polarity on the overall declining capacity of the EPS, the potassium ferricyanide trichloroacetic acid method outlined by Benzie and Strain was employed [47]. The measurements were conducted at a wavelength of 630 nm using a microtiter plate reader (Biotek ELX800; Biotek, Winooski, VT, USA). In the experimental setting, DMSO was the negative control, while ascorbic acid at 1 mg/ml was used as the positive control. The outcomes’ quantification was expressed in ascorbic acid equivalent (AAE) µg/mg of the EPS.
Anti-inflammatory human red blood cells and membrane stabilization (HRBCs-MSM) assay
A blood sample was taken from the author according to the research ethics committee (Ref. No. ERUFP-PM-23-001) from the Egyptian Russian University. The study of in vitro anti-inflammatory activity was conducted using the HRBCs-MSM method, following the protocol described by Anosike et al. [48].
Anticoagulant evaluation of the EPS by PT and PTT tests
Assays, PT, and APTT were tested: After combining citrated normal human plasma with a sample concentration solution, the mixture was incubated for 3 min at 37°C. Next, for PT testing, 0.20 ml of PT test reagent was added to the mix and pre-incubated for 3 min at 37°C, then the clotting time was noted. Similarly, 0.10 ml of PTT assay reagent was preincubated and added to the mixture under the same conditions afterward; 0.025 mol/l was preincubated for 3 min at 37°C, and the clotting time was recorded [49].
Wound healing assessment of EPS
Human normal skin fibroblasts, the HFB4 cell line, were obtained from the Holding Company for Biological Products and Vaccines (VACSERA) in Cairo, Egypt. The cells were seeded into six multi-well plates and allowed to grow until reaching confluency. At the start of the experiment, it was essential that all cell cultures had attained a confluent monolayer. A straight scratch was made using a yellow pipette tip to simulate a wound. To minimize the scratch breadth, we frequently produce the scratch with the pipette tip at an angle of about 30°. This enables imaging with the 10× objective of both wound edges [50].
Anti-lipase in vitro inhibition
Lipase stock solutions (1 mg/ml) were prepared in a 0.1 mM K3PO4 buffer (pH 6.0) and stored at –20°C. P-nitrophenyl butyrate (PNPB) was used to assess lipase inhibition activity. EPS at different concentrations (1.95–1000 µg/ml) and orlistat at comparable doses were pre-incubated with lipase for 1 h at 30°C in a potassium phosphate buffer to ascertain their lipase inhibitory action. Next, 0.1 µl of PNPB was added as a substrate to initiate the reaction at a final volume of 100 µl. The reaction’s release of p-nitrophenol was measured at 405 nm using a Biosystem 310-plus UV-visible spectrophotometer following a 5-minute incubation period at 30°C [51]. In addition, the negative control’s activity was evaluated both with and without the inhibitor.
EPS antidiabetic assessment
Anti-α-amylase testing
The α-amylase inhibition analysis was conducted by applying the 3,5-dinitrosalicylic acid (DNSA) method described by Wickramaratne et al. [52]. The concentrations of EPS ranged from 1.95 to 1000 µg/ml and were compared to the acarbose standard control, which also ranged from 1.95 to 1000 µg/ml. The absorbance measurements were taken at a wavelength of 540 nm using a UV-visible Biosystem 310 spectrophotometer. The IC50 values were derived from the graph by graphing the α-amylase inhibition % versus the concentration of EPS.
Anti-α-glucosidase examination
The methodology proposed by Pistia-Brueggeman and Hollingsworth (2001) was employed to evaluate the α-glucosidase inhibitory activity. The experimental EPS was tested at 1.95 to 1000 µg/ml concentrations. The results were then compared to those of the acarbose control, which also encompassed concentrations ranging from 1.95 to 1000 µg/ml. The absorbance measurements were conducted at a wavelength of 405 nm using a Biosystem 310 plus spectrophotometer. The IC50 values were calculated using a regression equation from graphing the doses tested against the enzyme inhibition [53].
Antimicrobial evaluation against G+ve and G-ve pathogenic bacteria
The antimicrobial effects of the EPS were evaluated using the agar well diffusion method against a range of bacterial strains from the ATCC collection. G+ve bacteria were Bacillus subtilis (ATCC 6633), Staphylococcus aureus (ATCC 6538) and Enterococcus faecalis (ATCC 29212). G-ve bacteria were Escherichia coli (ATCC 8739), Klebsiella pneumoniae (ATCC13883), and Salmonella typhi (ATCC 6539). The dried agar was smeared in three directions. Following a 15-minute drying period, an aseptic technique was employed to create a hole in the agar using a sterile cork borer with a diameter of 6–8 mm. Gentamicin was utilized as the control drug, and both gentamicin and EPSF8 were solubilized in DMSO at a concentration of 10 mg/ml. Subsequently, 100 units of EPSF8 were introduced into the well. The plates were incubated for 16–48 h immediately after disposal, and the widths of the inhibition zones surrounding the wells were measured to the nearest whole millimeter when there was a noticeable reduction in growth [54]. The investigation of minimum inhibitory concentrations (MICs) and minimum bactericidal concentrations (MBCs) was subsequently conducted under the guidelines set forth by the Clinical and Laboratory Standards Institute (CLSI) [55].
The anti-H. pylori activity was determined by the well agar diffusion method using Mueller Hinton agar plates containing 10% sheep blood. Wells were punched into the agar and filled with 100 µl of the antimicrobial agent solutions at desired concentrations. DMSO was used as the negative control. Positive controls were amoxicillin at 0.05 mg/ml, clarithromycin at 0.05 mg/ml, and metronidazole at 0.8 mg/ml. After 72 h of incubation at 37°C under microaerophilic conditions with humidity, the diameter of the inhibition zone around each antimicrobial agent well was measured and compared to the positive and negative controls.
Antibiofilm evaluation of the EPS
The impact of EPS on biofilm development was evaluated using 96-well polystyrene flat-bottom plates. To summarize, 300 µl of trypticase soy yeast broth (TSY) containing a final concentration of 106 CFU/ml was subjected to cultivation to 75%, 50%, and 25% of MBC of the previously tested organisms excluding E. coli. After 2 days of incubation at 37°C, the biofilm on the plates was dyed with 0.1% crystal violet aqueous solution for 15 min. After the incubation period, sterile dH2O was used to remove any residual stain from the plate. 250 µl of 95% C2H5OH was added to each well to dissolve the dye adhering to the cells. After 15 min, absorbance was measured at 570 nm using a microplate reader [56].
Results
Screening, identification, and phylogenetic identification of high EPS producing isolate
A comprehensive collection of 12 bacterial isolates derived from marine sediment samples from the Red Sea was obtained and submitted to a rigorous screening process to determine their ability to synthesize EPS. The screening procedure encompassed assessing cultural traits and morphological parameters and quantifying EPS production yield. The strain R9 of the marine bacterium exhibited the largest EPS yield (5.21 g/l). This EPS production predominantly comprised a major fraction, constituting 86.01% (three-volume ethanol). Microbiological analysis was conducted on the selected strain. The morphological and culture examination indicated a G+ve short rod that forms large, opaque white colonies with a rough and irregular surface morphology (Supplementary Table SI). The biochemical and physiological tests revealed a catalase-positive, starch-hydrolyzing bacterium that can reduce nitrate and ferment certain carbohydrates such as glucose and sucrose, but does not with maltose and lactose (Supplementary Table SII). Molecular 16srRNA sequencing followed, and the phylogenetic tree compared sequences that showed considerable similarity to the bacterium’s rRNA sequences. The acquired rRNA gene sequences matched Bacillus rugosus SYG20 (Figure 1), proving that the tree was assembled successfully. Accession number OR673614 confirmed Bacillus rugosus SYG20 identification. The DNA sequence was analyzed using BLAST and submitted to NCBI GenBank.

Structural and compositional analysis using UV, FT-IR, HPLC, uronic acid, and sulfate quantification
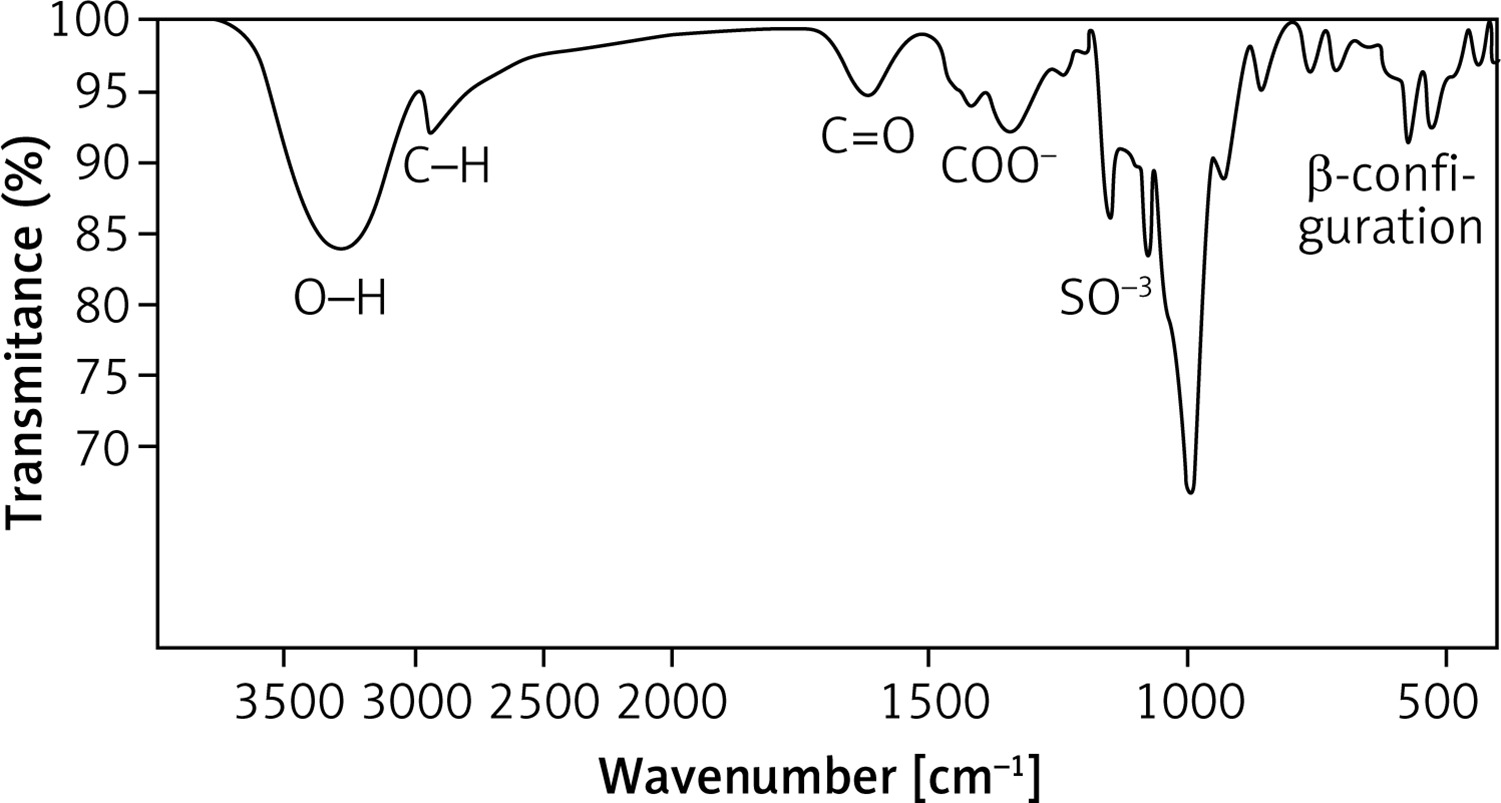
The Bacillus rugosus SYG20 strain was selected as the preferred candidate for producing exopolysaccharide (EPSR9), achieving a yield of 5.21 g/l. The unrefined residue underwent a purification process that included fractionation and precipitation. The EPSR9 sample was filtered through a membrane with a pore size of 100 microns after being treated with deionized water for 3 days. The EPSR9 that had undergone dialysis was treated with a progressive treatment with cold C2H5OH, resulting in fractional precipitation. Three ethanol precipitation procedures created the EPSR9 core fraction (86.01%) from crude EPS. The resulting fraction was then exposed to UV absorption spectra ranging from 200 to 800 nm (Supplementary Figure S1). EPSR9 had uronic acid (45.33%), sulfate (9.98%), and N-acetyl glucose amine (5.40%). As demonstrated by the FT-IR, the broad characteristic peak at 3275.03 cm–1 was assigned to OH–1 stretching vibration. The band at 2928.15 cm–1 corresponded to the sugar ring’s C-H stretching vibration. Also, absorption at 1632.68 cm–1 referred to COO– vibration and 1338.10 cm–1. The band at 1077.06 cm–1 indicated SO–3, and there was characteristic absorption at 860.28 cm–1 arising from β-configuration of the sugar units (Figure 2).
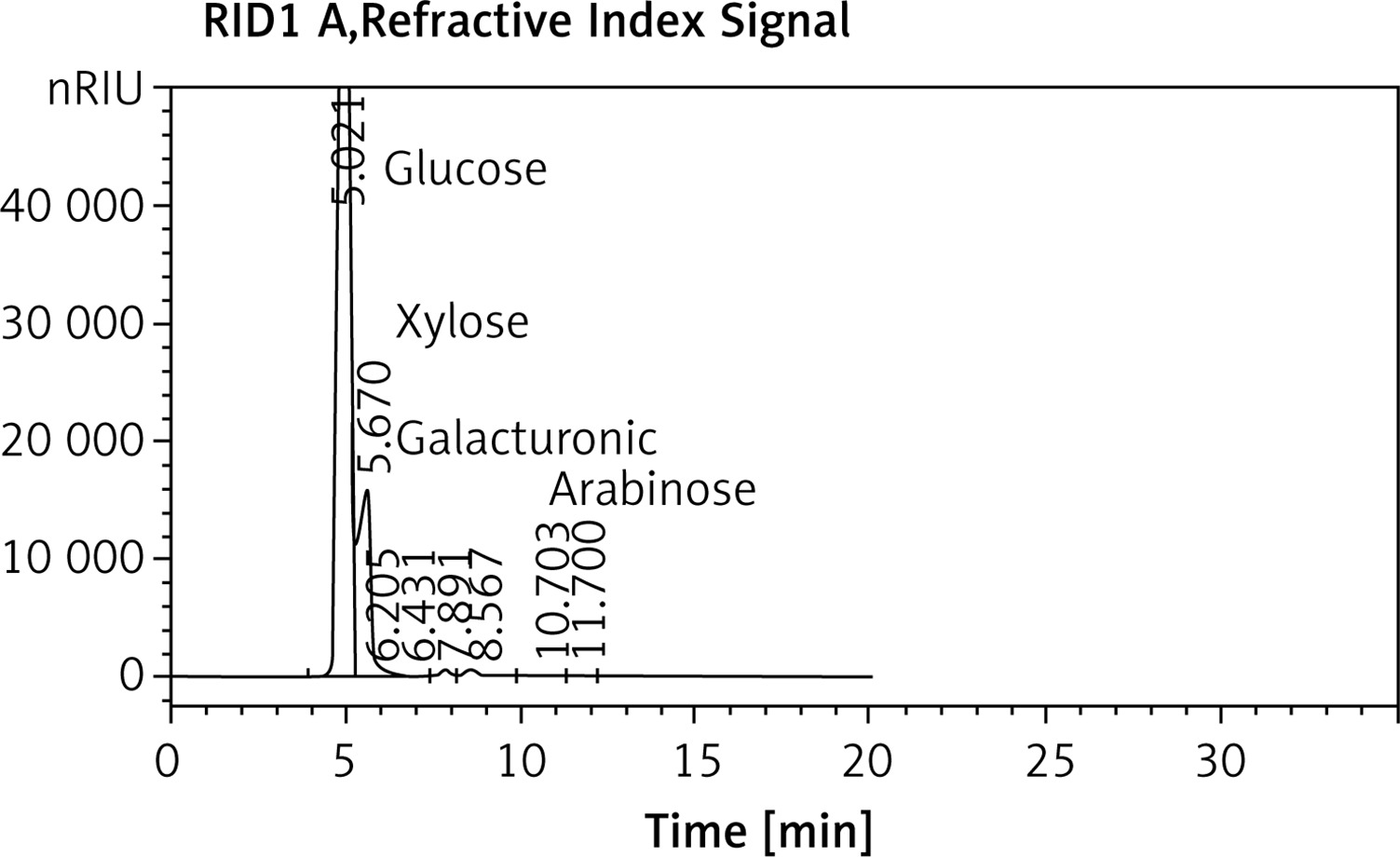
The HPLC chromatogram of EPSR9 revealed the monosaccharide fractions (glucose : xylose : galacturonic acid : arabinose) with molar ratio of 2 : 3 : 1 : 1, respectively (Figure 3).
Antioxidant evaluation of EPSR9 by DPPH, TAC and FRAP
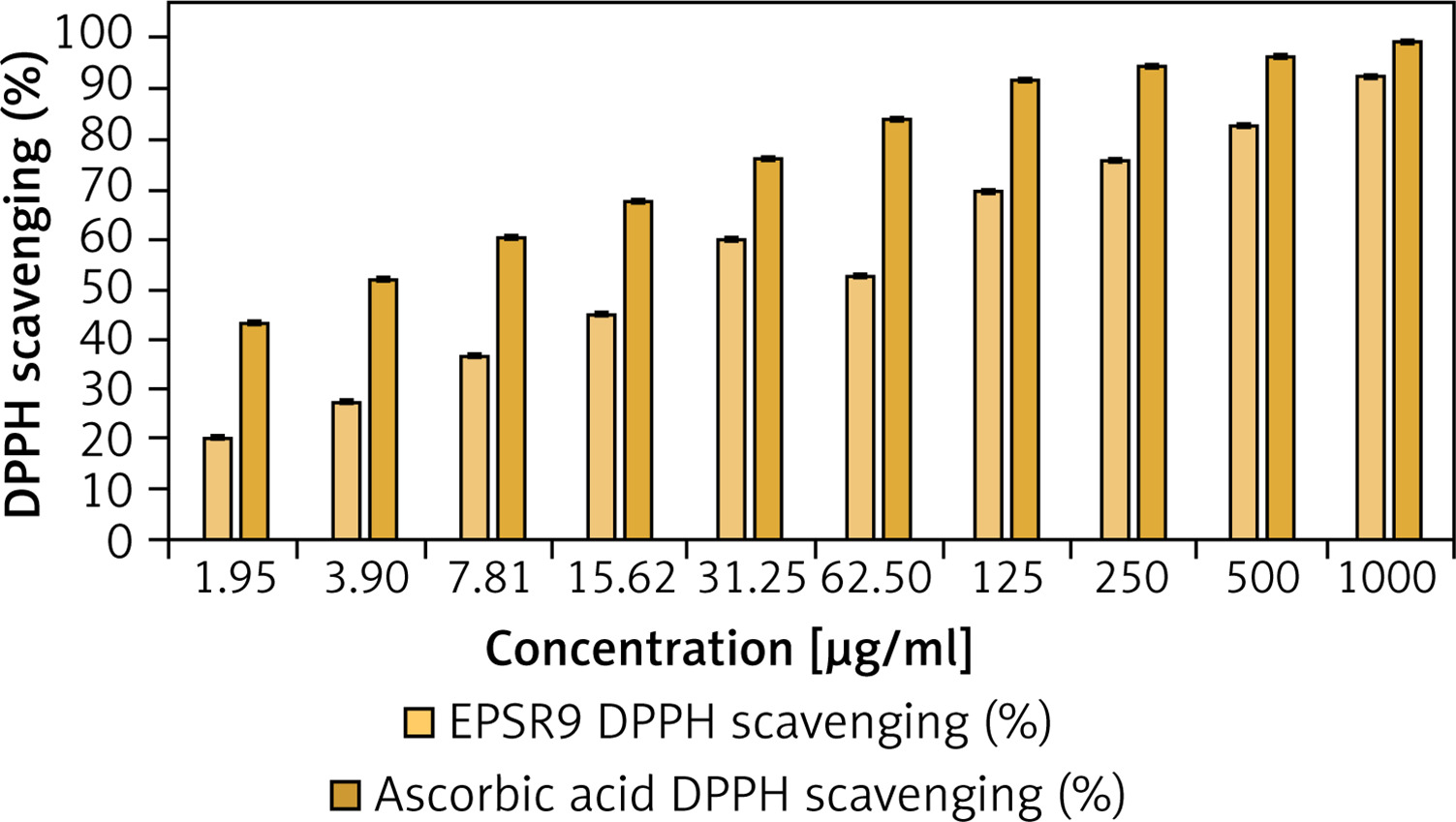
EPSR9 exhibited a noticeable dose-dependent and progressive increase in DPPH scavenging from 20.0% to 92.5% as the concentration increased from 1.95 to 1000 µg/ml across triplicate measurements. The IC50 value for EPSR9 was 25.6 ±0.001 µg/ml (Figure 4). The standard antioxidant ascorbic acid showed higher potency, with an IC50 of 2.52 ± 0.001 µg/ml (Supplementary Figure S2). Though EPSR9 displayed lower antioxidant activity than ascorbic acid, it still showed appreciable, dose-dependent radical scavenging capabilities. EPSR9 achieved 92.5% DPPH scavenging at the highest tested concentration compared to 99.3% for ascorbic acid.
Figure 4
EPSR9 (1.95 to 1000 μg/ml) DPPH radical scavenging % vs. ascorbic acid. Results presented as mean ± SE. One-way ANOVA (n = 3, p ≤ 0.05
Complementary assays comprehensively evaluated EPSR9’s antioxidant potential through different mechanisms and reaction environments. The TAC evaluation using the phosphomolybdenum method was conducted for EPSR9, resulting in a 417.77 AAE equivalent µg/ml (Table I). This was compared to the TAC ascorbic control (Supplementary Figure S2). EPSR9 was determined to possess a FRAP AAE equivalent concentration of 62.67 µg/ml, as indicated in Table II. This value was compared to the FRAP of the ascorbic acid standard, as reported in Supplementary Figure S3.
Anti-inflammatory assessment of EPSR9 by HRBC hemolytic and membrane stabilization assay
EPSR9 demonstrated significant in vitro anti-inflammatory effects in the HRBC hemolytic and membrane stabilization assay, evidenced by dose-dependent inhibition of hemolysis. EPSR9 at 100–1000 µg/ml concentrations progressively inhibited hypotonic solution-induced erythrocyte hemolysis from 81.8% to 99.0% (Table II). The standard anti-inflammatory drug indomethacin showed a dose-responsive reduction in HRBC lysis from 93.3% to 99.5% inhibition at 100-1000 µg/ml. At its highest tested concentration (1000 µg/ml), EPSR9 demonstrated comparable anti-inflammatory effects of 99% compared to 99.5% at the same tested concentration for indomethacin, preventing almost complete HRBC hemolysis.
Table II
Dose-responsive inhibition of HRBC hemolysis by EPSR9 and indomethacin
Dose-dependent anticoagulation by EPSR9 in coagulation screening tests
The exopolysaccharide EPSR9 exhibited dose-dependent anticoagulant activity in vitro as measured by PT and PTT assays. At 25–75 µg/ml concentrations, EPSR9 progressively extended PT from 18.7 to 49.3 s compared to 13 s for the standard control. Similarly, EPSR9 dose-dependently increased PTT from 33.5 to 60.3 s vs. 28 s for the typical control sample. The standard anticoagulant heparin showed greater potency, increasing PT to 22.5–99.8 s and PTT to 66.1–145.7 s at the same concentrations. While EPSR9 demonstrated lower anticoagulant effects than heparin, it still displayed significant dose-responsive antithrombotic activities in both assays (Table III).
In vitro wound healing potential of EPSR9 evidenced in the scratch assay
EPSR9 exhibited significant vitro wound healing activity compared to control cells in the scratch assay. The table presents the mean wound area measurements at different time points (0 h, 24 h, and 48 h) for EPSR9 and control cells. At the initial 0 h time point, the mean wound area was similar for both groups, with EPSR9 having a mean of 946.3333 µm2 and control cells having a mean of 941.6667 µm2. However, a significant difference emerged over 48 h. The EPSR9 group exhibited a remarkable reduction in the mean wound area, decreasing from 946.3333 µm2 at 0 h to 258.6667 µm2 at 48 h. This corresponds to a 72.66% wound closure rate for the EPSR9 group. In contrast, the control group showed a more modest reduction in the mean wound area, decreasing from 941.6667 µm2 at 0 h to 387.0000 µm2 at 48 h, corresponding to a 58.90% wound closure rate (Table IV, Figure 5).
Table IV
Effect of EPSR9 on in vitro scratch assay wound closure over 48 h

Anti-obesity evaluation of EPSR9 through lipase in vitro inhibition
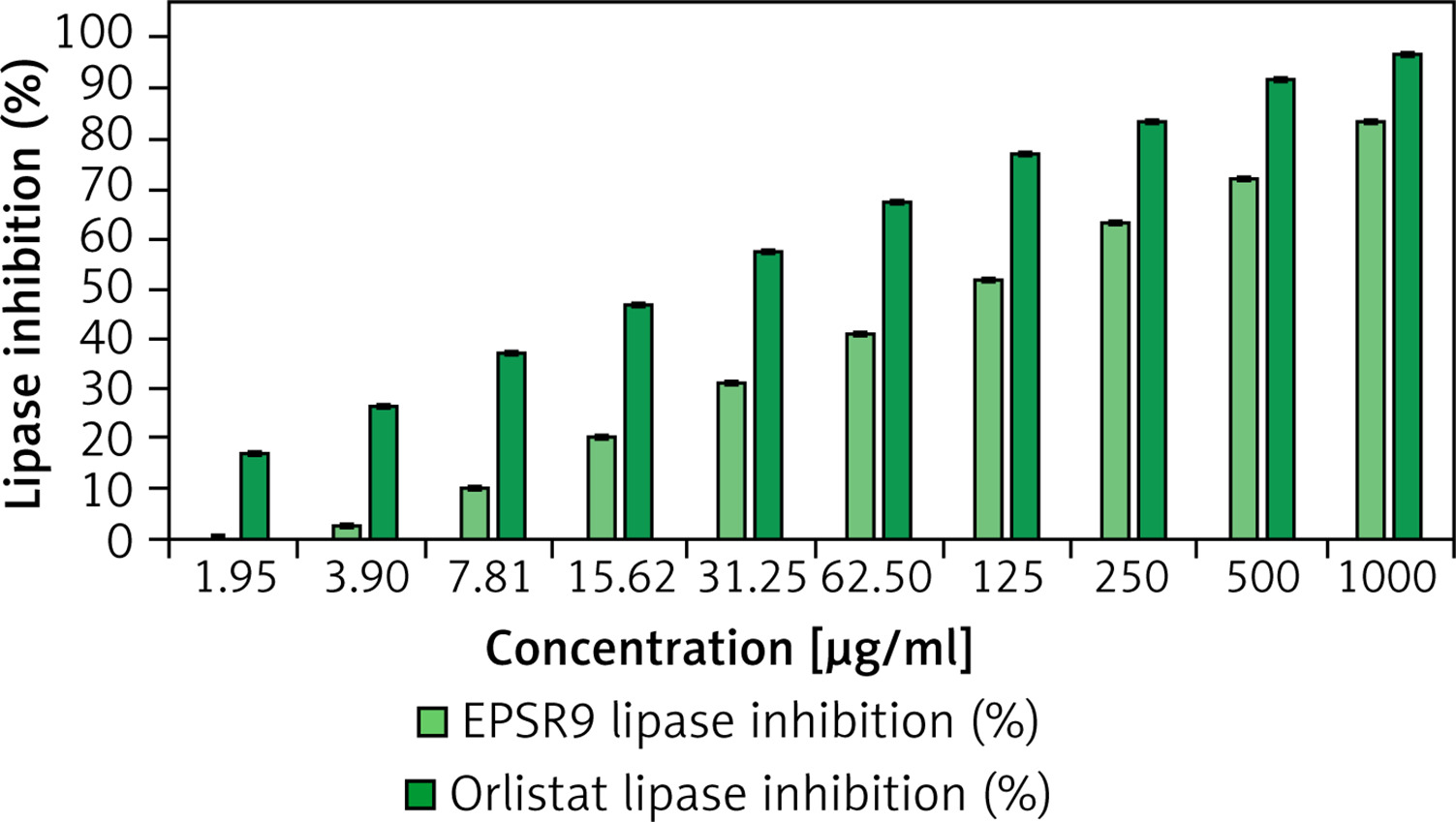
EPSR9 displayed concentration-dependent inhibition of lipase activity with an IC50 of 107.73 µg/ml (Figure 6), while orlistat had an IC50 of 20.08 µg/ml. At the highest tested concentration of 1000 µg/ml, EPSR9 inhibited 83.8% lipase activity compared to 96.8% inhibition by orlistat.
Antidiabetic in vitro inhibitory investigation of EPSR9
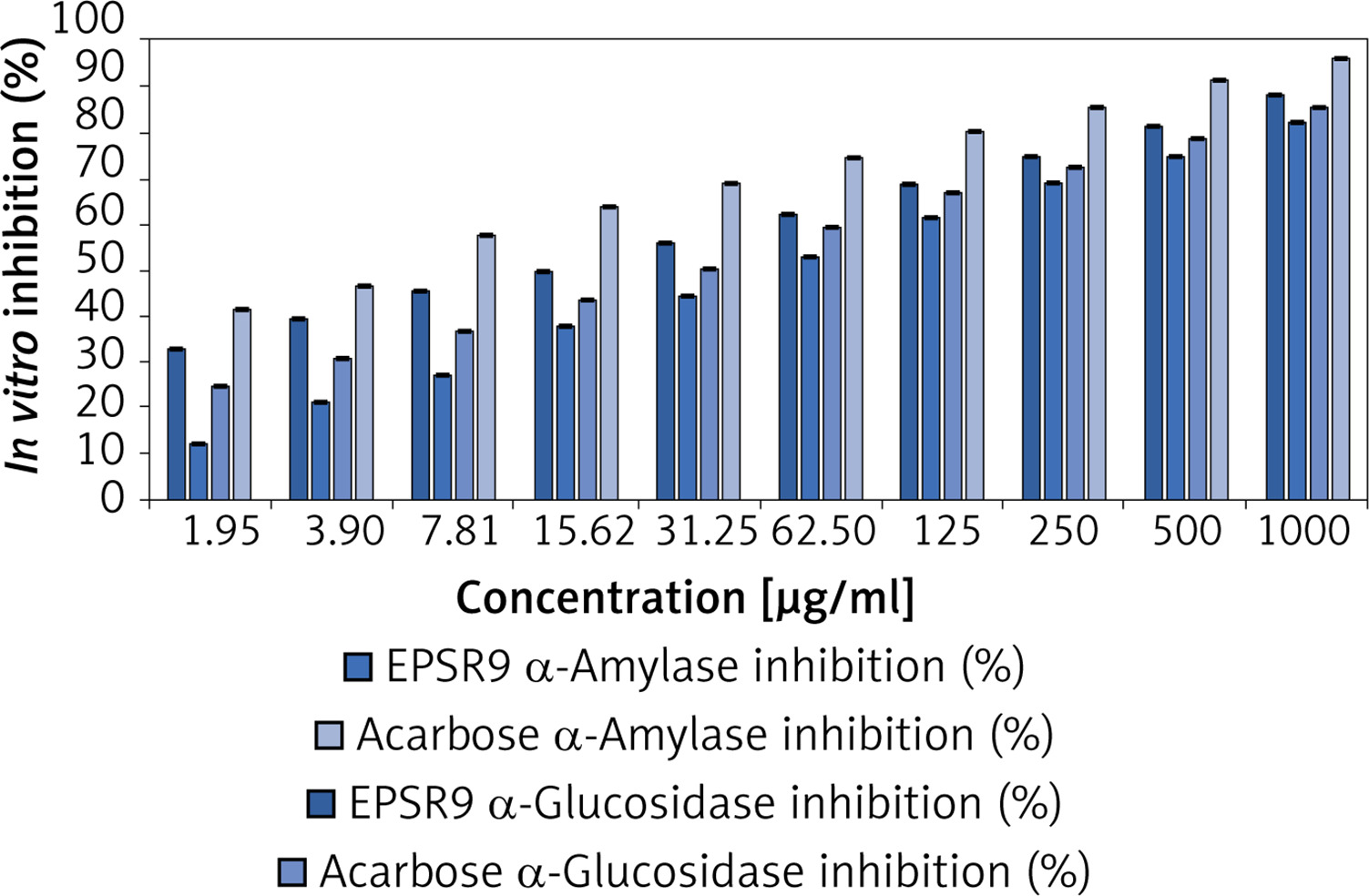
The experiment tested the in vitro inhibitory effects of the marine bacterial polysaccharide EPSR9 on α-amylase and α-glucosidase, two key enzymes involved in carbohydrate digestion and implicated in type 2 diabetes, in comparison to the standard drug acarbose. EPSR9 exhibited concentration-dependent inhibition of both α-amylase (IC50 14.37 µg/ml) and α-glucosidase (IC50 26.73 µg/ml) (Figure 7). At the maximum tested concentration of 1000 µg/ml, EPSR9 inhibited α-amylase and α-glucosidase by 88.2% and 85.3%, respectively, compared to 82.1% and 96.1% for acarbose at the same test concentration. In contrast, the standard acarbose displayed IC50 values of 50.93 µg/ml and 4.13 µg/ml for α-amylase and α-glucosidase inhibition, respectively.
Antimicrobial screening of EPS
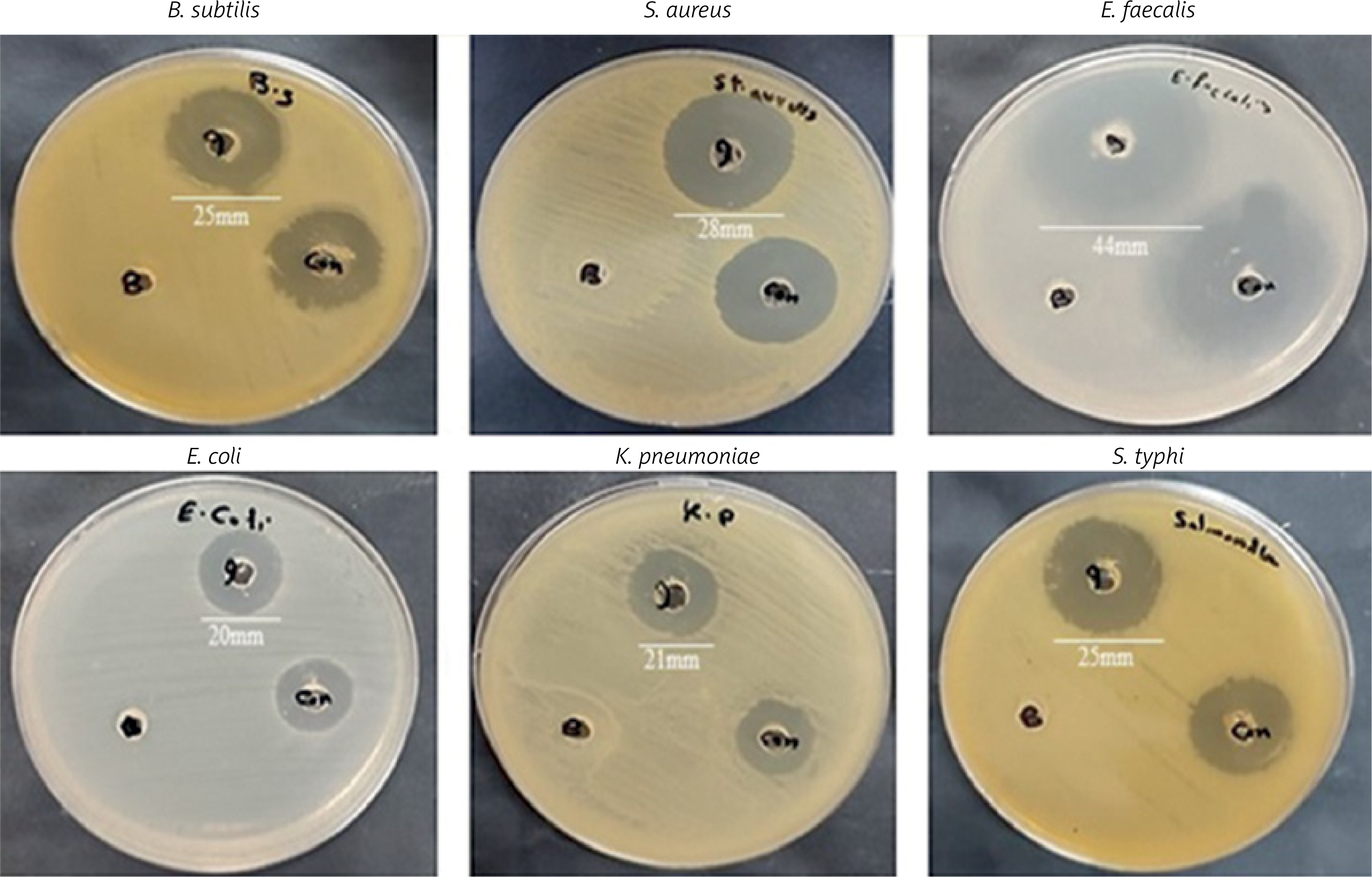
EPSR9 exhibited bactericidal activity against Gram-positive and Gram-negative bacteria, although it was more potent against the Gram-positive strains tested (Figure 8). Against the G-ve bacteria tested, EPSR9 showed moderate inhibitory activity. Against E. coli, it had an inhibition zone of 20 mm compared to 16 mm for gentamicin and MIC and MBC values of 125 and 250 µg/ml, respectively, giving an MBC/MIC ratio of 2, indicating a bactericidal effect. Against K. pneumoniae, the inhibition zone was 21 mm vs. 17 mm for gentamicin, with high MIC and MBC values of 250 and 500 µg/ml and an MBC/MIC ratio of 1, indicating bactericidal potential. EPSR9 inhibited Salmonella typhi with an inhibition zone of 25 mm compared to 24 mm by gentamicin, and MIC and MBC of 31.25 and 62.5 µg/ml, respectively, with an MBC/MIC ratio of 2, confirming bactericidal action (Table V, Figure 9).
Table V
Antibacterial potential of EPSR9 against G+ve and G-ve bacterial pathogens
Figure 8
The antibacterial effect of EPSR9 is represented as inhibition zones (mm) against ATCC G+ve and G-ve bacteria
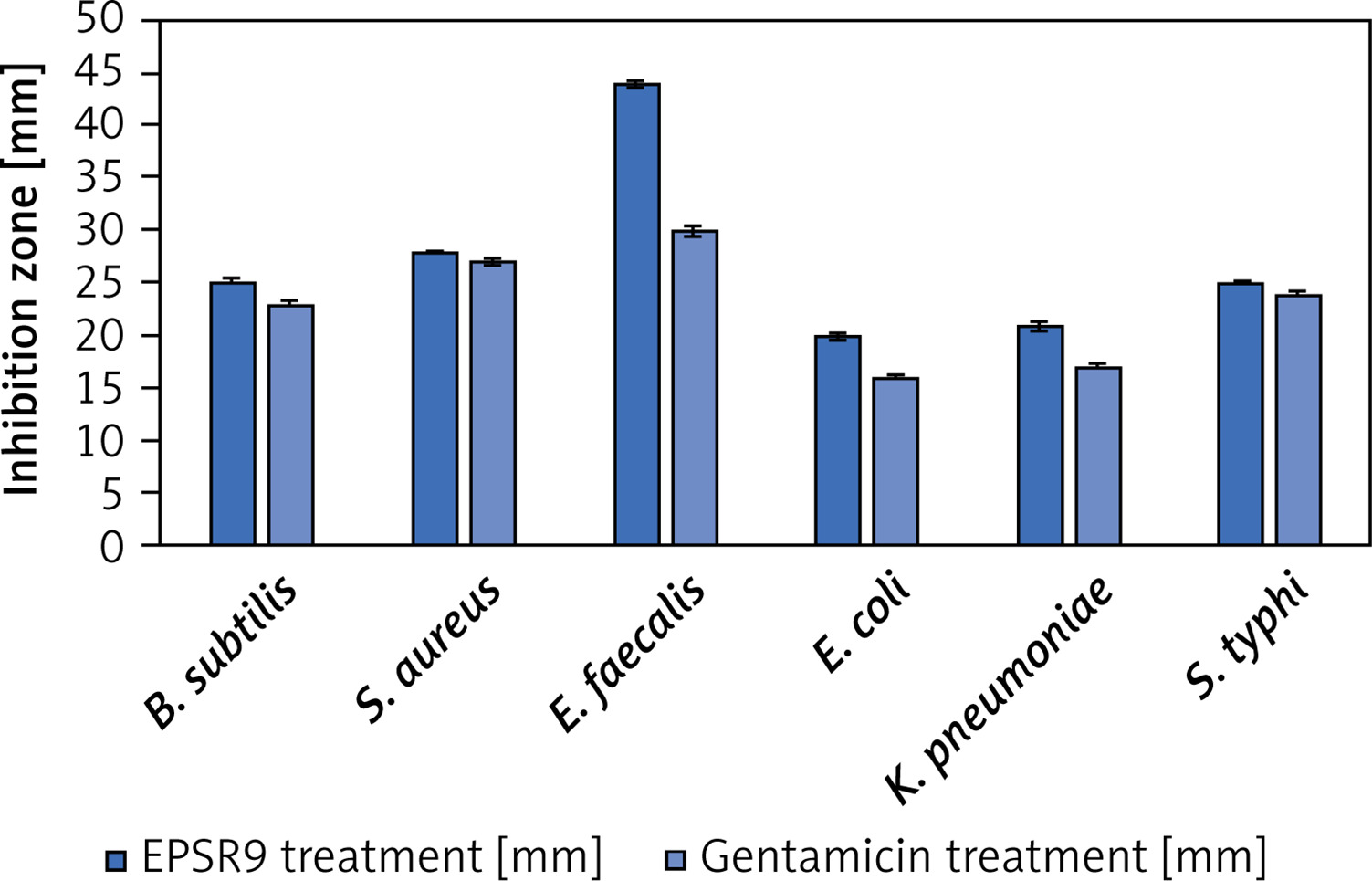
Against the G+ve species, EPSR9 exhibited stronger antimicrobial properties. It inhibited B. subtilis with a zone of 25 mm vs 23 mm for gentamicin and MIC and MBC values of 31.25 and 62.5 µg/ml, giving a bactericidal MBC/MIC ratio of 2. Against S. aureus, the inhibition zone was 28 mm for EPSR9 and 27 mm for gentamicin, with MIC of 62.5 µg/ml and MBC of 125 µg/ml, also showing a bactericidal effect (MBC/MIC ratio 2). EPSR9 displayed potent activity against E. faecalis, with a 44 mm inhibition zone compared to 30 mm for gentamicin and very low MIC and MBC values of 3.9 and 7.8 µg/ml, respectively, confirming bactericidal potential via the MBC/MIC ratio of 2 (Table V, Figure 9).
EPSR9 demonstrated more potent antimicrobial effects against gastric ulcer pathogenic Helicobacter pylori. It formed a larger inhibition zone of 24.67 mm compared to 21.00 mm for the positive control (amoxicillin at 0.05 mg/ml, clarithromycin at 0.05 mg/ml, and metronidazole at 0.8 mg/ml). EPSR9 also had a lower MIC of 31.25 µg/ml versus 62.5 µg/ml for the control. Both compounds showed an MBC of 62.5 µg/ml; however, EPSR9’s MBC/MIC index was lower at 2. EPSR9 exhibited promising antibacterial activity against H. pylori in vitro, as evidenced by a larger inhibition zone, lower MIC, and comparable MBC to the positive control. Additionally, the anti-biofilm activity of different (sub-MBCs) of the EPSR9 against H. pylori biofilms was further tested. At 25% of the MBC, EPSR9 inhibited 89.51% of H. pylori biofilm formation. This activity increased to 92.75% inhibition at 50% of the MBC and 95.60% at 75% of the MBC (Table VI).
Table VI
Antibacterial represented as inhibition zone (mm) and anti-biofilm activity at different MBC% of EPSR9 against Helicobacter pylori
| Inhibition zone [mm] | MIC [µg/ml] | MBC [µg/ml] | MBC/MIC Index | |
|---|---|---|---|---|
| EPSR9 | 24.67 | 31.25 | 62.5 | 2 |
| Positive control | 21.00 | 62.5 | 62.5 | 1 |
Following this, the anti-biofilm activity of EPSR9 at various % MBC concentrations was tested against the same tested bacterial strains but not E. coli (Figure 10). First, the antibiofilm of G+ve bacteria was investigated. According to the findings, S. aureus showed the highest level of 67.28% anti-biofilm activity at 75% MBC. Meanwhile, at the lowest measured percentage of 25% MBC, its minimal activity was recorded at 37.06%. Additionally, Enterococcus faecalis showed a similar pattern, with its highest level of inhibition reaching 84.83% at 75% MBC (Supplementary Table SIII). Meanwhile, at 25% MBC, its activity dropped to 61.76%. Lastly, Bacillus subtilis was investigated and found to be most vulnerable at 75% MBC, where a remarkable 86.08% anti-biofilm effect occurred. However, when the concentration was reduced to 25% MBC, this dropped to 61.18% (Table VII).
Table VII
EPSR9 antibiofilm activity against B. subtilis and S. typhi at 25, 50, and 75% MBC
Figure 10
Antibiofilm activity of EPSR9 against different G+ve and G-ve ATCC bacteria at different % MBC
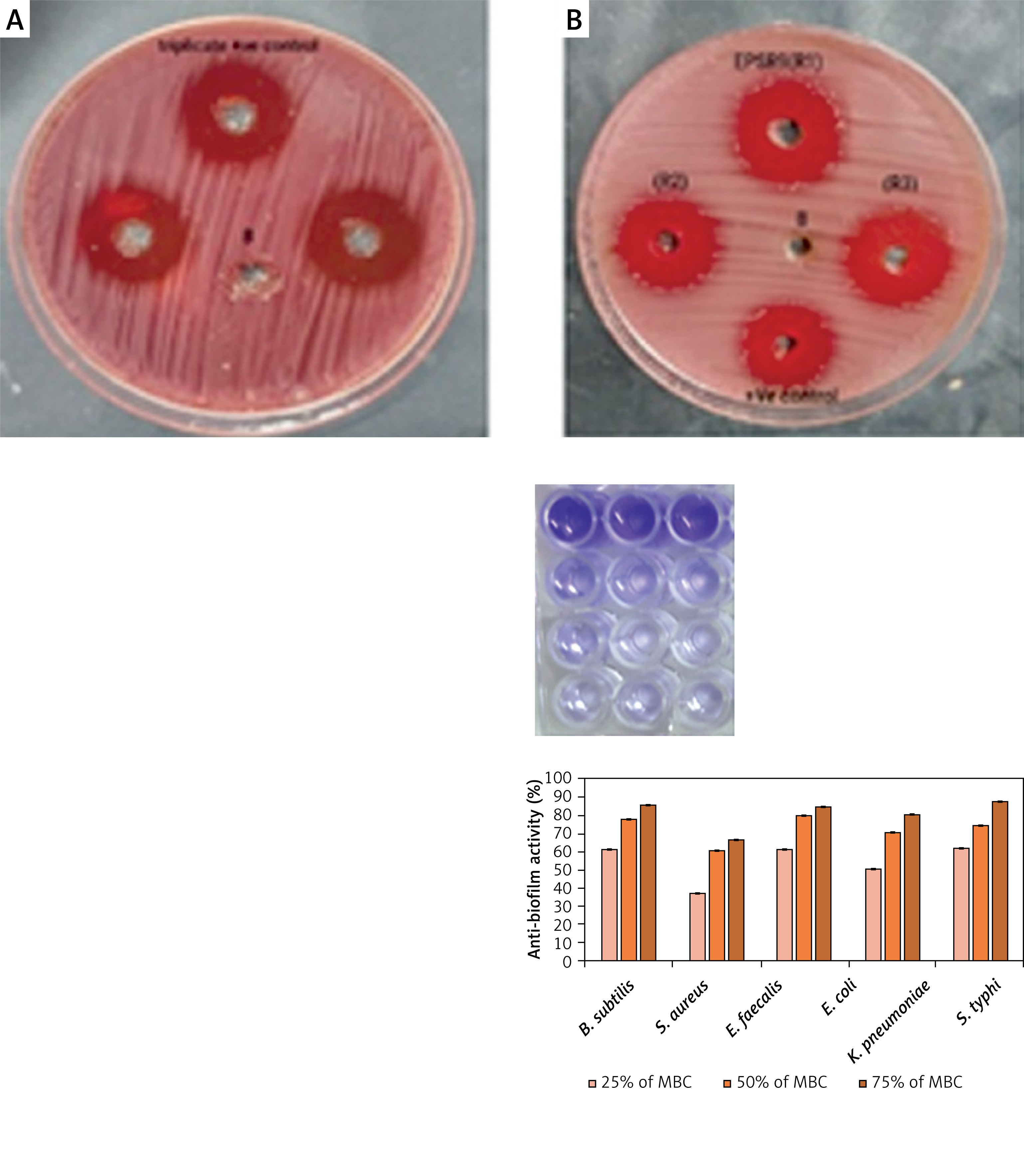
Next, the evaluation of G-ve bacteria was conducted. Initially, Klebsiella pneumoniae showed 80.84% optimum activity at 75% MBC. At 25% MBC, this dropped to the smaller but still considerable value of 50.86% (Supplementary Table SIII). Next, Salmonella typhi showed an even higher maximal inhibition of 88.05% at 75% MBC. Under less concentrated settings, it was less effective, declining to 61.90% at 25% MBC (Table VII). In general, EPSR9 outperformed all other pathogens at a maximum of 75% MBC. It also suppressed Gram-negative organisms more powerfully than Gram-positive ones. Essential insights into EPSR9’s concentration-dependent anti-biofilm ability against a variety of therapeutically relevant bacteria were obtained from this thorough investigation. This comprehensive investigation emphasized the importance of EPSR9’s concentration-dependent anti-biofilm abilities against pathogenic bacteria.
Discussion
Twelve bacterial isolates were isolated from Red Sea marine sand samples. These isolates were tested for exopolysaccharide biosynthesis. Marine bacterium strain R9 yielded the highest amounts of EPS (5.21 g/l). Microbiological investigation revealed a G+ve, short rod-shaped bacterium with large white colonies (Supplementary Table SI). Biochemical analyses indicated that it hydrolyzes starch, ferments carbohydrates, and is catalase-positive (Supplementary Table SII). 16S rRNA sequencing with accession number OR673614 identified it as Bacillus rugosus SYG20, validated by NCBI GenBank gene sequence matching (Figure 1).
Exopolysaccharide (EPSR9) has been extracted from Bacillus rugosus SYG20 because of its high yield (5.21 g/l) and core fraction (86.01%) (three volume C2H5OH). EPSR9 was shown to have uronic acid (45.33%), sulfate (9.98%), and N-acetyl glucose amine (5.40%) using FT-IR spectroscopy (Figure 2). EPSR9’s monosaccharide fractions consisted of glucose, xylose, galacturonic acid, and arabinose in the following molar ratio by HPLC analysis: 2 : 3 : 1 : 1 (Figure 3).
Starting with antioxidant screening of EPSR9 by DPPH, TAC, and FRAP, as the concentration increased from 1.95 to 1000 µg/ml, EPSR9 demonstrated dose-dependent and progressive DPPH radical scavenging from 20.0% to 92.5%, achieving 92.5% at the maximum concentration compared to 99.3% for ascorbic acid. The IC50 value was 25.6 ±0.001 µg/ml, while ascorbic acid was 2.52 ±0.001 µg/ml (Figure 4). The TAC result utilizing the phosphomolybdenum technique was 417.77 AAE µg/ml, while the FRAP value for EPSR9 was 62.67 AAE µg/ml, compared to routine ascorbic acid (Table I).
The antioxidative capacities of microbial EPSs have been observed to be substantial. The structural elements of monosaccharides are categorized as reducing sugars due to their possession of aldoses and ketoses or their ability to undergo interconversion between these two forms. In addition, such capacity could be attributed to the diverse array of functional groups, such as -OH, -COOH, -CONH2, -SO42-, -SH, -COCH3, -C=O, and many more. These functional groups exhibit the ability to donate electron pairs, undergo proton loss, or aid the process of metal binding. Consequently, free radicals become stable molecules [5, 57]. Furthermore, it has been hypothesized that negatively charged functional groups could generate an acidic environment, enhancing the hydrolysis of EPSs. Therefore, the augmentation of antioxidant activity is facilitated by a higher level of exposed hemiacetal hydroxyl groups [58].
Next, EPSR9 was tested as an anti-inflammatory natural compound by HRBCs-MSM assay. EPSR9 reduced inflammation. The dose-dependent suppression of hypotonic solution-induced erythrocyte hemolysis ranged from 81.8% to 99.0% at doses of 100–1000 µg/ml (Table II). Indomethacin, a common anti-inflammatory medication, reduced HRBC lysis dose responsively from 93.3% to 99.5% at 100–1000 µg/ml. In the maximum concentration of 1000 µg/ml, EPSR9 showed similar anti-inflammatory effects to indomethacin (99% vs. 99.5% inhibition), almost totally preventing hemolysis.
The anticoagulant property of EPSR9 was dose-dependent in vitro as evaluated by PT and PTT assays. EPSR9 increased PT from 18.7 to 49.3 s at 25–75 µg/ml, compared to 13 s for heparin. EPSR9 also dose-dependently elevated PTT from 33.5 to 60.3 s vs. 28 s for heparin. Heparin increased PT to 22.5–99.8 s and PTT to 66.1–145.7 s at the same doses. Although EPSR9 had less effective anticoagulant effects than heparin, it showed dose-responsive antithrombotic activity in both assays (Table III).
The scratch experiment showed that EPSR9 had substantial wound healing activity relative to control cells. EPSR9 treatment reduced the wound area from 946.3333 µm2 to 258.667 µm2 in 48 h, resulting in 72.66% closure. In comparison, control cells showed 58.90% wound closure, dropping from 941.66 µm2 to 387 µm2 (Table IV, Figure 5). Recent research has indicated that EPSs generated by certain marine bacteria can promote wound healing by stimulating fibroblast and keratinocyte migration and proliferation, documenting the bioactivities of such molecules and their potential usage as wound-care products [28, 59, 60]. Synechocystis aquatilis Sauvageau B90.79 synthesized sulfated EPS that acted as an anticoagulant and a complement modulator [61]. Anticoagulant action was observed in the EPS derived from Alteromonas infernus after chemical modifications involving sulfation and depolymerization [62]. Shirzad et al. documented the anti-elastase, anticollagenase, antioxidant, and wound-healing properties of EPSs synthesized by certain strains of Lactobacillus. These EPSs can potentially be developed into suitable agents for combating skin aging [63].
Moving to the anti-obesity evaluation of EPSR9 through lipase in vitro inhibition, EPSR9 showed concentration-dependent inhibition of lipase activity with an IC50 of 107.73 µg/ml. In comparison, orlistat had an IC50 of 20.08 µg/ml. At 1000 µg/ml, EPSR9 inhibited 83.8% lipase activity compared to 96.8% inhibition by orlistat (Figure 6). The EPSs generated by Lactobacillus plantarum GA06 and GA11 exhibited in vitro cholesterol reduction efficiencies of 36.7% and 28.6%, respectively. The observed EPSs showed a notable capacity for cholesterol binding [64]. Another in vitro study of an EPS (EPS400) from Limosilactobacillus fermentum NCDC400 exhibited a significant cholesterol-lowering efficacy of 90.32% [65]. One notable characteristic of the EPS generated by Leuconostoc mesenteroides LM187 is its considerable capacity to reduce cholesterol levels, with an efficacy rate of 53% [66].
Next, EPSR9 was compared to acarbose for its inhibitory effects on α-amylase and α-glucosidase. EPSR9 inhibited α-amylase (IC50 14.37 µg/ml) and α-glucosidase (IC50 26.73 µg/ml) concentration-dependently at 1000 µg/ml; EPSR9 inhibited the enzymes by 88.2% and 85.3%, compared to 82.1% and 96.1% for acarbose (Figure 7). Acarbose’s IC50 values were 50.93 µg/ml and 4.13 µg/ml for both enzymes. EPSR9 exhibits a modest inhibitory effect against carbohydrate-digesting enzymes in vitro and requires additional investigation in diabetes animal models to understand its antidiabetic efficacy and mechanism of action.
The antidiabetic property is regarded as one of the activities demonstrated by microbial EPSs, which may be measured by evaluating the inhibition of α-amylase and α-glucosidase enzymes. This inhibitory mechanism that impedes the hydrolysis of glucose confers advantages to individuals with diabetes. Following our findings, EPS derived from Enterococcus faecium MS79 exhibited 91% and 92% inhibitory activity against α-amylase and α-glucosidase, respectively [67]. EPSs derived from marine cyanobacteria have been reported to have antidiabetic properties via blocking α-glucosidase. Pseudanabaena and Chroococcus sp. extracted EPS reduced α-glucosidase activity by 14.02% and 13.00%, respectively [68].
The mechanism by which EPS inhibits α-amylase and α-glucosidase is not clearly understood. EPS appears to inhibit hydrolysis via binding to the active site of enzymes or substrates. Another hypothesis is that EPS reduces glucose levels by activating insulin receptors and increasing glucose utilization [69]. The hypoglycemic influence of EPS could also be attributed to its ability to stimulate Langerhans islets and enhance peripheral sensitivity to residual insulin, and its antioxidant potency [70, 71].
Then, EPSR9 was tested against a spectrum of G+ve and G-ve pathogenic ATCC bacteria. EPSR9’s effectiveness against G+ve bacteria was more pronounced (Figures 8, 9). Inhibition zones for E. coli were 20 mm for EPSR9 and 16 mm for gentamicin; corresponding MIC/MBC values were 125/250 µg/ml. Zones against K. pneumoniae were 21 mm compared to 17 mm, and the MIC and MBC were 250 and 500 µg/ml, respectively. S. typhi zones were 25 mm vs. 24 mm; MIC/MBC 32.25/62.5 µg/ml. The B. subtilis zones measured 25 vs 23 mm, with a MIC/MBC of 31.25/62.5 µg/ml. The S. aureus zones measured 28 mm vs. 27 mm, with a MIC/MBC of 62.5/125 µg/ml. With a zone of 44 mm vs. 30 mm, EPSR9 significantly inhibited E. faecalis; MIC/MBC 3.9/7.8 µg/ml (Table V). EPSR9 showed improved efficacy against Gram-positive bacteria, substantially suppressing E. faecalis. The evidence highlights the intriguing antibacterial potential of EPSR9 and calls for more research into it as a cutting-edge antibacterial drug.
Against H. pylori, EPSR9 showed more potent antibacterial activity. Compared to the positive control, which had an inhibition zone of 21.00 mm, it generated a larger one of 24.67 mm (Table VI). Additionally, EPSR9’s MIC was lower, 31.25 µg/ml, than the control’s, which was 62.5 µg/ml. EPSR9’s MBC/MIC value was lower at 2; however, both compounds displayed an MBC of 62.5 µg/ml. EPSR9 had encouraging antibacterial activity against H. pylori in vitro, as shown by an extended inhibitory zone, a lower MIC, and an MBC similar to the positive control.
EPSR9’s efficacy as an anti-biofilm agent was tested against the same bacterial spectrum except for E. coli (Figure 10). When tested against Gram-positive bacteria, S. aureus had the highest activity: 67.28% at 75% MBC, decreasing at 25% MBC to 37.06% (Supplementary Table SIII). The highest percentage of E. faecalis was 84.83% at 75% MBC, and the lowest rate was 61.76% at 25% MBC (Supplementary Table SIII). The most vulnerable strain was B. subtilis, which declined to 61.18% at 25% MBC from 86.08% at 75% MBC (Table VII). Within G-ve, K. pneumoniae did the best, with an 80.84% highest inhibition rate at 75% MBC and a 50.86% lowest antibiofilm rate at 25% MBC (Supplementary Table SIII). At 75% MBC, S. typhi was inhibited up to 88.05%, but it decreased to 61.90% at 25% MBC. In general, EPSR9 was more effective against Gram-negative bacteria than Gram-positive bacteria at 75% MBC (Table VII).
Sub-MBCs of EPSR9 were then examined for their ability to inhibit H. pylori biofilms. At 25% of the MBC, EPSR9 exhibited strong anti-biofilm efficacy, preventing 89.51% of H. pylori biofilm formation. At 75% of the MBC, the anti-biofilm activity reached 95.60% inhibition after increasing in a dose-dependent manner. EPSR9 exhibited antibacterial solid and anti-biofilm properties against H. pylori at concentrations lower than its MBC, and at 75% of MBC, it nearly completely (95.60%) inhibited biofilm formation (Table VI).
The antibacterial responses of microbial EPSs are potentially associated with disrupting the structural integrity of bacterial cell membranes, cell walls, and respiratory chains, hence impacting the machinery involved in cell division [72, 73]. Microbial EPSs cannot permeate cell membranes, thus exerting their antibacterial effects, likely through their interaction with oligopeptides or acyl-homoserine lactones in G+ve and G-ve bacteria.
The chemicals mentioned above are signal molecules associated with biofilm formation. Cell communication disruption and biofilm development suppression are observed due to EPSs acting through this mechanism [74]. Hence, it is plausible to consider such EPSs as potentially efficacious therapeutic agents for mitigating chronic and recurrent infections associated with biofilms.
Moreover, EPSs have been observed to diminish the autoaggregation of bacterial pathogens, rendering them more vulnerable to the immune response within the host [75]. Additionally, EPSs can adhere to microbial pathogens using their EPS. The coaggregation process enhances these entities’ antibacterial potential by obstructing the receptors or channels on G-ve pathogenic bacteria’s outer membrane [76]. EPSs exhibit a diverse array of functional groups, encompassing hydroxyl, phosphate, and carbonyl units. Therefore, it has been proposed that these functional groups play a role in interaction of bacterial pathogens with their cell walls or membranes. Such interaction may elucidate the antimicrobial attributes [77].
Formation of biofilms is intricately associated with the colonization and dissemination of pathogenic bacteria. These factors significantly influence the virulence of pathogens, intercellular communication processes, and many infection states. A potential in vitro mechanism behind the observed antibiofilm action of EPSR9 derived by Bacillus rugosus SYG20 could involve the disruption of cell-to-cell communication. This disruption may occur through the binding of EPSR9 to glycocalyx receptors located on the surface of pathogenic bacteria or interfering with the signal molecules associated with biofilm formation. Consequently, this generation of biofilms is impeded, leading to the eventual exertion of the antimicrobial effect.
The molecular structure of EPSR9 needs to be verified, and further research is required to investigate the compound’s biocompatibility in vivo, its precise mode of action, and whether or not it can alter the composition of the gut microbiome. Further, genetic engineering strategies such as mutagenic strains and gene alterations can expand the number of marine bacterial strains that biosynthesize valuable EPSs with unique structures and bioactivities to achieve increased EPS yields.
In conclusion, the present investigation isolated, extracted, and characterized a bioactive bacterial exopolysaccharide (EPSR9) synthesized by the marine bacterium Bacillus rugosus SYG20 and comprehensively demonstrated its remarkable pharmaceutical potential through extensive in vitro assessments. EPSR9 exhibited potent antioxidant, anti-inflammatory, and anticoagulant properties and significant wound healing, anti-obesity, and antidiabetic activities. Notably, EPSR9 displayed broad-spectrum bactericidal effects against Gram-positive and Gram-negative pathogens, including Helicobacter pylori, and exhibited potent anti-biofilm activity, effectively disrupting the formation of bacterial biofilms. These comprehensive findings underscore the immense therapeutic potential of this marine-derived exopolysaccharide, which could serve as a valuable natural compound for developing multifunctional therapeutic agents.