Introduction
Among several inflammatory autoimmune disorders rheumatoid arthritis (RA) is one of the most common, characterized by synovial membrane inflammation and immune cell-mediated joint destruction. It primarily affects the joints, resulting in painful swollen joints to severe polyarthritis with progressive articular cartilage destruction. The worldwide prevalence of RA is about 0.24% [1]. According to Cross et al., RA continues to remain a source of modest global infirmity, with serious consequences in affected individuals. Collected data from studies suggested that the disease occurrence is inconsistent in different racial groups [2]. In Pakistan, disease prevalence is still unspecified, but it has been perceived that it is more common in the country’s northern parts than the southern part [3]. It depends on the combination of various environmental as well as a number of genetic factors [4]. It has also been observed that RA occurs in individuals having multiple common genetic factors and these genetic risk factors are expected with 60% heritability [5]. Multiple genetic risk factors of various candidate genes incline the patient towards disease susceptibility by the development of different clinical symptoms following exposure to unknown environmental factors [6]. Microsatellite mapping across the HLA region suggested that a nearby class III region possibly contributes to disease susceptibility or severity [7–9]. Evidence emphasized that within the class III region the tumor necrosis factor-α (TNF-α) gene is a major candidate for RA [10].
Single nucleotide polymorphisms (SNPs) in the promoter regions are likely to have a potential to cause differential expression of proteins and possibly have an association with disease [11]. TNF gene promoter polymorphisms may influence the transcriptional activity by transforming the transcription factor binding site [12]. Various SNPs have been reported for the TNF gene promoter region. Among them SNP at –863 position is involved in NF-κB binding affecting the transcriptional regulation [13]. In HLA-DR4+ individuals TNF –863A is found to be prognostic for severe joint disease [14]. Moreover, some studies have also found a positive association of TNF –863A allele and RA outcome [14]. Conversely, inconsistency among the different studies exists regarding TNF-α promoter polymorphisms’ association with RA outcome [14, 15]. There are limited data available concerning SNPs and RA disease susceptibility association in Pakistan [16, 17]. Still, there is a great need to explore the association studies of RA susceptible genes’ polymorphism not only in our study group but also in different ethnic groups due to conflicting results. Thus, considering the above statements, in this study we further evaluated the TNF-α –863C/A variant in Pakistani patients with RA to assess any potential association.
Material and methods
The study included 268 human subjects. Among them 134 individuals were patients with RA and 134 individuals were ethnically matched controls. In this study, the physical parameters of the RA patients in a Pakistani population were amassed to investigate any association of these clinical features with the patients. The percentage of males and females and the average age of all the included individuals in the study were calculated for the analysis of SNP. All the included patients were diagnosed by a rheumatologist from Rehmat Noor Clinic, Rawalpindi, working in collaboration with our institution.
Inclusion criteria: patients included in this study fulfilled the American College of Rheumatology (ACR) criteria 2011 for RA diagnosis and classification. Exclusion criteria: individuals overlapping with any other rheumatic or autoimmune disease were excluded from the study. To diagnose RA different inflammatory markers including erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), rheumatoid factor (RF) and anti-cyclic citrullinated peptide antibody (anti-CCP) were used. Anti-CCP of all the patients was analyzed by the fully automated chemiluminescent immunoassay system COBAS ELECSYS 411. Reference values used for detection of markers were: anti-CCP (negative < 17 U/ml; positive > 17 U/ml), RF (up to 20 IU/ml), C-reactive protein (normal range < 6 mg/l), ESR (normal range for males: 0–9 mm; for females: 0–15 mm).
Research was carried out in compliance with the Helsinki Declaration. Written informed consent was obtained from the entire study group. The study was approved by the ethical committee of the Institutional Review Board (IRB) of ASAB-NUST.
Clinical features of RA patients and healthy individuals collected are shown in Table I.
Table I
Clinical characteristics of patients (n = 134)
Sampling and DNA extraction
Blood samples of all the enrolled individuals were collected in an ethylenediaminetetraacetic acid (EDTA) vacutainer. The samples were immediately dispatched to the Functional Genomics and Immuno-genetics laboratory (IGL) of Atta-ur-Rahman School of Applied Biosciences (ASAB), National University of Sciences and Technology (NUST), Islamabad, Pakistan. From the blood cells of collected EDTA blood samples genomic deoxyribonucleic acid (DNA) was extracted using the organic method. The extracted DNA was qualitatively analyzed on 1% agarose gel and quantitatively analyzed by a bio-photometer. The extracted DNA was then stored at 4°C before any further processing.
Design of primers
SNP selected for the association study was located in the promoter region of the TNF-α gene. The sequence of the TNF-α promoter region was taken from National Centre for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/) with the accession number rs1800630. Forward and reverse primers for SNP were then manually designed and their properties were calculated by the oligonucleotide properties calculator Oligo- Calc [18]. The specificity was checked on Primer-BLAST. One common forward (TNF-F-5’TGTGTGTGTGTGTCTGGGAGTGAGAA3’) and two reverse (TNF-R1-5’TCTACATGGCCCTGTCTTCGTTAAGG3’ and TNF-R2-5’TCTACATGGCCCTGTCTTCGTTAAGT3’) primers resulting in a 389 bp fragment were designed for studying the TNFA –863A>C polymorphism. The prerequisite Internal Amplification Control forward (IAC-F-5’ATGGTCTTAGTATAGCTTGCAGCCTTGT3’) and reverse (IAC-R-5’TGCAGATACCATCATCCTGGCTTCAAG3’) primers resulting in a 144 bp fragment were also designed considering β-globin as the standard in order to validate the reaction.
Genotyping
The RA patients and healthy individuals were examined for the occurrence of the polymorphism by allele specific ARMS-PCR. Denatured DNA templates at 95°C for 5 min were hybridized with primers at 54°C for 45 s, subsequently amplified in a 96-well thermocycler 2,720 (Applied Biosystems) with 35 cycles of PCR. The PCR product and loading dye (0.25% bromophenol blue in 40% sucrose solution) was mixed carefully for loading the mixture in the wells of 2% (w/v) agarose gel. The gel was ethidium bromide stained. After running gel electrophoresis at 120 V in 1XTBE buffer for about 0.5 h, the gel was analyzed on the Dolphin-Doc plus gel documentation system (Wealtech). Length of the PCR product was determined by comparing the PCR product size with a 50 bp DNA ladder.
Statistical analysis
Data were statistically examined for any association of a variant with disease by GraphPad Prism 6 software. Hardy-Weinberg equilibrium (HWE) of genotypes was confirmed by the χ2 test (1 df). All binomial variables were assessed by χ2/Fisher exact test for association analysis of the variant with disease in our study group. To estimate the degree of association of each allele, the odds ratio (OR) and its 95% confidence interval (CI) were calculated.
Results
The study included a total of 268 individuals. In the case group 86.86% were female and 13.14% were male, with a mean age of 44.15 ±12.21 years, and in the control group 80.59% were female and 19.40% were male, with a mean age of 35.74 ±7.1 years. Out of 134 patients, 58 (43.3%) individuals were > 45 years of age while 76 (56.7%) patients were ≤ 45 years of age. All the 134 patients were positive for the anti-CCP test and 133 patients were positive for the RF factor.
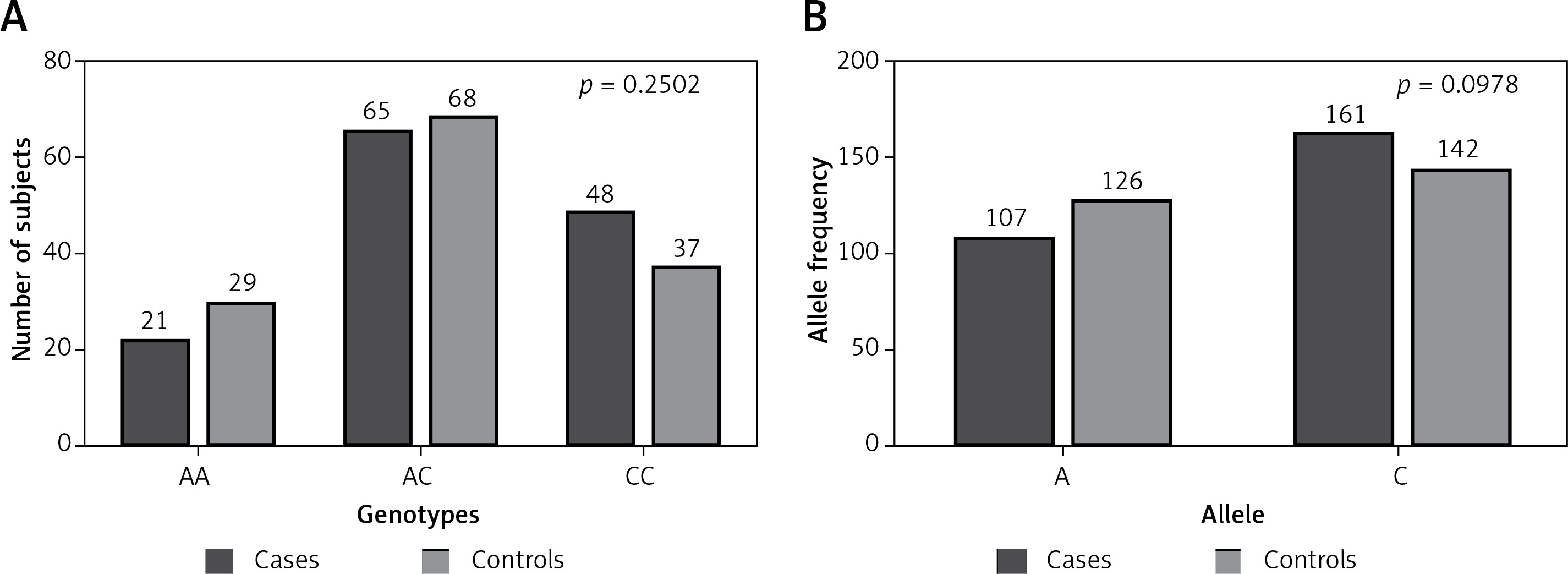
All possible allele combinations distributed in our subjects were studied. The allele counting method for TNF-α –863C/A (rs1800630) was employed to compute the allele and genotype frequencies, and the polymorphism was verified for any deviation from Hardy-Weinberg equilibrium (HWE). Both sets were suitable for auxiliary examination and association studies. The p-value estimated for RA patients’ was 0.8968 with χ2 = 0.02, while the p-value was 0.8299 with χ2 = 0.05 for the control group. Genotype and allele frequency distribution calculated and matched with the frequency of arbitrary healthy people (Figure 1). The value of association was found to be 2.771 with the probability of error (p-value) of 0.2502 (Table II). Allele frequency distribution considered using two-tailed analysis shows no substantial difference between cases and controls with the odds ratio (95% CI) of 0.7490 (0.5317–1.055).
Table II
Distribution of TNF-α –863C/A (rs1800630) genotypes and alleles in cases and controls
Figure 1
Distribution of different genotypes and alleles of TNF-α gene promoter –863C/A (rs1800630) polymorphism among our study groups (cases and controls). A – p = 0.2502 with χ2 = 2.771; B – p = 0.0978 with χ2 = 2.741. Calculation using χ2/Fisher exact test shows no statistically significant difference
Discussion
Rheumatoid arthritis is an inflammatory autoimmune disease affecting humans throughout the world that causes joint inflammation, swelling and pain. A wide variety of factors together with environmental and genetic elements contribute to the disease pathogenesis and progression. Genome scanning indicated a strong genetic component [19], revealing that there is more than one region that is linked to the disease [20–22]. Cumulative studies also counsel that patients whose genetic history includes several common elements are more susceptible to RA. Genetic susceptibility contributed by these genetic risk elements or alleles was estimated to be approximately 30% [23]. According to our investigation in a Pakistani populace the rate of occurrence of RA is higher in females than in males. In our study, we found that a higher percentage of females (86.86%) were affected than males (13.14%), which is consistent with findings from Taiwan [24] and a North Indian cohort [25], implying that gender may have a substantial consequence for RA susceptibility. Studies show that promoter regions of RA candidate genes are extremely polymorphic, which has been accompanied with disease susceptibility as well as severity in different populations [26]. Among them, TNF-α plays a central role in disease pathogenesis, increased levels having been reported in inflamed joints [27]. High genetic variability with several SNPs has been determined in the promoter area of the TNF-α gene [28]. These SNPs could possibly alter transcription factor binding sites, affecting promoter activity and ultimately leading to altered mRNA and protein levels [29]. TNF-α is biologically active in two distinctly different forms, i.e., soluble form (solTNF) and transmembrane form (tmTNF). Both forms are specified for their roles; solTNF has an imperative, perhaps the dominant, role in the inflammatory response, while tmTNF plays a fundamental role in maintaining innate immunity against infections [30]. It is a multifunctional pro-inflammatory cytokine that is involved in different pathological processes including autoimmunity and neurodegenerative diseases [31].
Studies have focused on the association of TNF-α gene polymorphisms with RA. The role of TNF-α in pathogenesis of autoimmune diseases has been discussed in a number of studies [11, 32]. Despite some contradictory views, so far available data reveal that the variants of the TNF gene have the potential for disease progression and could act as potential genetic risk factors [33, 34]. Despite this, auxiliary studies are needed to ascertain whether the formerly recognized TNF-α gene promoter variations act as potential markers of RA. TNF-α gene polymorphisms variants have been found associated with several autoimmune diseases [35-37]inflammatory arthritis like psoriatic arthritis (PsA) [38-41]juvenile rheumatoid arthritis [42], and systemic juvenile rheumatoid arthritis [43]. In some studies, TNF-α promoter variants were found with aggressive disease [44]. According to Skoog et al., TNF-α –863 variant rs1800630 was found to be associated with the disease severity. A SNP at position –863 is involved in NF-κB binding affecting the transcriptional regulation [13]. Tumor necrosis factor –863A allele lessens the NF-κB p50/p50 binding that directs the enhanced TNF production in human monocytes [12]. Moreover, some studies found a positive association of TNF –863A allele and RA outcome [14]. In HLA-DR4+ individuals, TNF –863A is found to be prognostic for severe joint disease [14]. In our study the frequency of allele distribution at position –863 of TNF-α was comparable with a North Indian population in a previous report [15], but different from that of Udalova et al. [12]. TNF-α –863A allele frequency distribution revealed no significant difference between our study groups when our RA patients (39.93%) were compared with the control group (47.01%) with p-values of 0.0978 and 0.7490 (95% CI: 0.5317–1.055), which is consistent with the North Indian population with no association of TNF-α –863 with the disease. Results with lack of a positive association were also consistent with the findings of Uglialoro et al. [45]. In our study groups, TNF-α –863A minor allele frequency was comparable with Japanese data [46]. Our data do not support the TNF-α –863A allele as a genetic component contributing to RA disease susceptibility in our population as the distribution of the TNF-α –863A allele was not different between the study groups. However, these allelic polymorphisms explain comparatively incomplete clinical intervention as RA implicates multiple genes. Thus, to describe the relationship of TNF-α alleles with RA, independent larger population study validation is required to confirm the association.
In conclusion, in our study group, the TNF-α –863A allele was not found to be a genetic risk susceptibility element in RA progression. Nonetheless, information related to TNF promoter variants is imperative to gain a better insight into RA genetics. However, in order to endorse and extend the results, further investigations on larger populations are required.