Introduction
Rheumatoid arthritis (RA) is a common autoimmune heterogeneous joint disease of still unknown etiology. In RA inflammation starts in the synovial membrane and spreads further to surrounding tissues, leading finally to cartilage and bone destruction [1, 2]. Various genetic [3, 4] and epigenetic [5] factors have been found to be associated with RA, but also environmental factors such as cigarette smoke, and viral and bacterial infections play a significant role in development of the disease [6, 7]. The pathogenesis of RA is regulated by T- and B-cells. B-cells are involved in the production of autoantibodies such as rheumatoid factor (RF) [8] and anti-cyclic citrullinated peptide autoantibodies (ACPA) [9, 10]. T-cells produce a variety of pro-inflammatory cytokines, especially tumor necrosis factor (TNF), interleukin 6 (IL-6) as well as vascular endothelial growth factor (VEGF), which are involved in degradation of bone and cartilage within the joint [11]. The pathogenesis of the disease may also be related to IL-33. Extracellular IL-33 is a critical enhancer of TNF-induced RA synovial fibroblast activation [12]. IL-33 in experimental models of collagen-induced arthritis exhibits triggering properties, and the administration of interleukin 33 intensifies the disease process [13, 14].
One of the characteristic features of RA is oxidative stress [15], most likely induced and stimulated by inappropriate B- and T-cell activity. It is associated with a higher level of reactive oxygen species (ROS) production in RA patients as compared with healthy subjects. The main factor that increases the release of ROS in RA patients is overproduction of TNF [16]. TNF controls cell proliferation or cell death. The altered level of nuclear factor kappa-B (NF-κB) increases TNF-induced death, concurrent with sustained Jun N-terminal kinase (JNK) activation via the death response. JNK activation in NF-κB-deficient cells depends on the ROS, but the influence of ROS in stimulation of JNK is still unclear [17]. Moreover, TNF-α, by increasing the expression of arginase in endothelial cells during ischemia/reperfusion (I/R) injury, leads to a decrease in the availability of l-arginine for nitric oxidase synthase, thereby increasing the production of O2−•. Overproduction of O2−• impairs the NO-mediated vasodilation and favors endothelial dysfunction. TNF-α inhibitors significantly reduce inflammation in patients with chronic autoimmune diseases, improving endothelial function and reducing the risk of acute cardiovascular complications [18, 19]. ROS also contribute to the depolymerisation of hyaluronic acid, causing increased bone resorption and reduction of joint viscosity. The elevated ROS level was correlated with various clinical parameters of RA such as seropositive or seronegative status and DAS score [20].
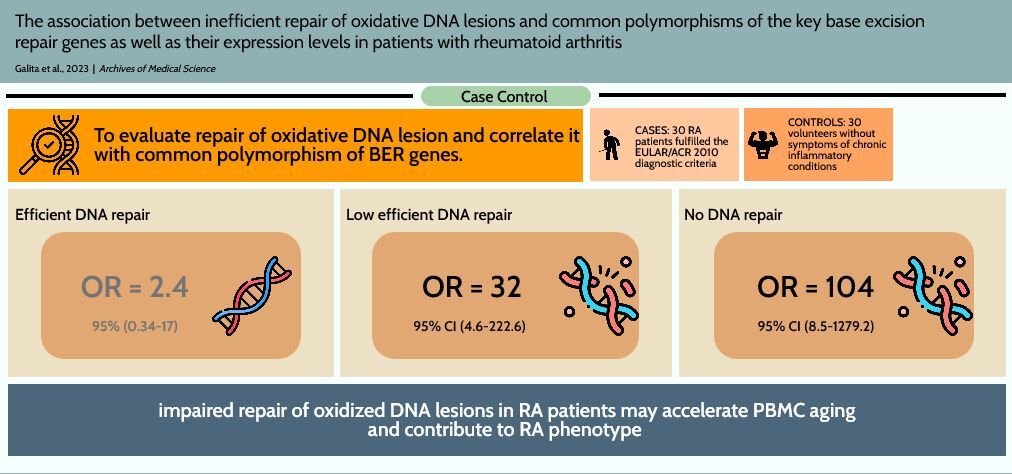
The negative ROS effect in RA is not limited to joints. Excessive ROS production results in accumulations of oxidative DNA lesions in peripheral blood mononuclear cells (PBMCs) isolated from RA patients, as we demonstrated previously [21]. We have hypothesized that oxidative stress together with an impaired DNA damage response (DDR) to oxidative DNA lesions (limited to base excision repair (BER) pathway) could be responsible for increased incidence in RA patients of some diseases with a genetic instability background such as lymphoma and lung cancer [22]. Therefore, the aim of this study was to evaluate repair of oxidative DNA lesions and correlate it with common polymorphism of BER genes as well as their expression level, as the results from our previous studies suggest that polymorphism of DNA repair genes may have a phenotypic effects in repair of oxidative DNA lesions [23–26]. Therefore, we determined the level of oxidative DNA lesions and the kinetics of repair of DNA damage induced by tert-butyl hydroperoxide (TBH) in PBMCs of 30 RA patients and 30 healthy individuals. The metrics from the DNA damage and repair study were correlated with the genotypes of common polymorphisms of the key BER genes as well as their expression levels. DNA damage and repair were evaluated by alkaline single cell gel electrophoresis (comet assay), the genotypes of the polymorphism were determined by TaqMan SNP Genotyping Assay, and PrimeTime qPCR Assay was used to analyze the expression profile of genes related to BER.
Material and methods
Patients
The study group included 30 patients with RA (22 women and 8 men; mean age: 59.3 ±15 years) selected from patients of the Department of Rheumatology and outpatient clinic and 30 volunteers (24 women and 6 men; mean age: 61.5 ±11.3) recruited from patients without symptoms of chronic inflammatory conditions. Such a number of subjects provides sufficient statistical power (> 0.80) for final conclusions. This cohort study has been approved by the Institutional Bioethics Committee of the Medical University of Lodz (Lodz, Poland) (no. RNN/07/18/KE, approval date: 16 January 2018). All RA patients fulfilled the EULAR/ACR 2010 diagnostic criteria of RA. Persons with past or present malignancy in first degree family members as a potential reason for DNA instability were excluded from the study.
Methods
Sample collection and preparation
Blood samples were collected from both patients and controls into anticoagulant tubes with a capacity up to 9 ml. Next, PBMC were isolated as well as DNA and RNA. Total RNA was isolated from peripheral blood using the RiboPure RNA Purification Kit according to the manufacturer’s protocol. Isolated RNA was transcribed into cDNA to a final concentration of 100 ng using GoScript Reverse Transcriptase according to the manufacturer’s protocol. Genomic DNA was prepared using the GeneMatrix Blood DNA purification Kit (EURx, Gdansk, Poland) according to the manufacturer’s protocol.
Analysis of SNPs
Genotypes were determined using TaqMan SNP Genotyping Assay. We analyzed ten SNPs in XRCC1, OGG1, UNG, MBD4, MUTYH, TDG, SMUG1 genes (respectively rs25487, rs1052133, rs246079, rs151095402, rs2307293, rs3219472, rs3219489, rs3219493, rs4135054 and rs3087404). The PCR reaction (total volume 25 µl) including 5 µl of 5x HOT FIREPol Probe qPCR Mix (Solis), 1 µl of DNA (100 ng), 1.25 µl 20x TaqMan SNP primers and 17.75 µl of RNase-free water conditions were as follows: polymerase activation (10 min, 95°C), 30 cycles of denaturation (15 s, 95°C) annealing/extension (60s, 60°C). Genotype determination was made in Bio-Rad CFX96 system (Bio-Rad).
We performed PrimeTime qPCR Assay (Integrated DNA Technologies) to analyze the expression profile of 15 genes associated with BER: OGG1 (Hs.PT.58.38536541 and Hs.PT.58.38797078) MUTYH (Hs.PT.58.4052997 and Hs.PT.58.38885205), NTHL1 (Hs.PT.58.45671999), NEIL1 (Hs.PT.58.38858353), NEIL2 (Hs.PT.58.39722884), UNG (Hs.PT.58.39999493), TDG (Hs.PT.58.1836292.g), LIG3 (Hs.PT.58.4393909), PARP1 (Hs.PT.56a.112995), PARP3 (Hs.PT.58.39766379 and Hs.PT.58.40275253) MPG (Hs.PT.58.19216181), APEX1 (Hs.PT.56a.3182919), APEX2 (Hs.PT.58.26859287), MBD4 (Hs.PT.58.39947182), NEIL3 (Hs.PT.58.1086590) and SMUG1 (Hs.PT.58.27762894). We used 2 reference genes: ACTB (Hs.PT.39a.22214847) and RPLP0 (Hs.PT.39a.22214824). The expression of selected reference genes was not sensitive to inflammation [27, 28]. The total reaction volume was 10 µl, including: 1 µl of cDNA, 1 µl of primers, 2 µl 5x HOT FIREPol Probe qPCR Mix (Solis) and 6 µl of nuclease free water. Condition for the reaction were suggested by the manufacturer of the HOT FIREPol Probe qPCR Mix: 15-minute enzyme activation at 95°C, 40 cycles of denaturation (10 s, 95°C), annealing/extension of 60 s, 60°C. qPCR reaction was conducted in the Bio-Rad CFX96 system (BioRad). Gene expression was calculated in relation to that of the reference genes (ΔCt sample = Ct target gene – Ct reference gene). Subsequently, the relative mRNA expression was calculated as fold = 2–ΔCt sample. Expression status was determined from the data regarding the expression value of the analyzed gene in the control patient population. Based on these results, 3 groups were defined. Samples with results above or equal to the median value (3rd and 4th quartiles) were classified as normal expression (Group 1), those located below the median in quartile 2 as low expression (Group 2), and in quartile 1 as very low expression/no expression (Group 3).
Analysis of efficacy of DNA repair
The alkaline version of the comet assay with modifications [21] was used to evaluate the efficacy of DNA repair. The comet assay is a rapid, sensitive and selective technique for quantifying and analyzing DNA damage in individual cells. The DNA damaging agent selected for this study was TBH. TBH is a well-known agent that induces oxidative DNA lesions repaired mainly by BER pathway [29]. We used TBH at 7 µM for 15 min on ice – this concentration did not affect the viability of PMBCs (viability of PMBCs was always > 80%). The kinetics of DNA repair was calculated by estimation of the level of DNA damage induced by TBH during the 60 min of the repair process. The DNA damage measured immediately after exposure to TBH was set as 100% of DNA damage to avoid any problems associated with individual and RA-driven variation in TBH sensitivity. Next, we measured the percentage of repaired DNA after 60 min. By subtracting the obtained value from 100, we obtained the individual DNA repair efficiency (DReff, the lower the value, the better). Next, the DNA repair ranks of individuals were determined similarly to expression analysis. All subjects were divided into four groups regarding quartiles for individual DNA repair efficiency of the control group. Quartile 1 means highly efficient DNA repair, quartile 2 efficient repair, quartile 3 low efficient DNA repair, and quartile 4 no repair.
Global methylation analysis
Global methylation was assessed using the MethylFlash Global DNA Methylation (5-mC) ELISA Easy Kit. In this assay, DNA is applied to the wells of the plate, which have a high affinity for DNA after treatment. The methylated DNA fraction is recognized using capture and detection antibodies and then quantified colorimetrically by reading the absorbance on a microplate spectrophotometer. 100 ng of isolated DNA from patients with rheumatoid arthritis and controls were applied to the well strips included in the kit. The analysis procedure was performed according to the manufacturer’s protocol.
Statistical analysis
Descriptive data were expressed as mean and standard deviations (SD). The data from the comet assay are presented as the median ± range. The normal distribution of continuous variables was confirmed by the Shapiro-Wilk test. The Mann-Whitney rank sum test was used to compare DNA damage between patients with RA and healthy controls, and it was decided based on the normality test. The relationships among the continuous variables were assessed using Spearman’s rank correlation coefficient. Multinomial logistic regression analyses were performed to calculate odds ratios (ORs) and 95% confidence intervals (CIs) for the effects of DNA repair status and other variables on RA. All variables included in the final multivariate models were determined to be independent by assessing their collinearity. Age, sex, genotypes and expression level of BER genes were included as independent variables in univariate and multivariate multinomial logistic regression analyses. Only matching variables and factors that altered the ORs by 10% were included in the final multivariate models. The quality of the models was determined by the Hosmer-Lemeshow test. All statistical analyses were performed with TIBCO Statistica 13.3 (Palo Alto, CA 94304, USA). In all tests, a p-value < 0.05 was used.
Results
Characteristics of the study population
There were no significant differences in the distributions of age, sex and smoking status between cases and controls. Mean disease duration was 12.27 ±10.7 years (from 1 to 37 years). Eighteen patients were currently (for at least 1 month before blood collection) treated with methotrexate (MTX), 3 patients with sulfasalazine, and 9 patients did not receive disease-modifying anti-rheumatic drugs (DMARDs) within the last month. Glucocorticosteroids (GCS) were used for treatment of 16 patients. Five patients did not have rheumatoid factor levels (positive in 25 cases). In addition, we determined the C-reactive protein (CRP) level (18.7 ±22.60 g/dl). The disease activity was also assessed based on the Disease Activity Score in 28 joints with CRP (DAS28-CRP). Remission was defined as a DAS28 score < 1.7 (2 patients), low disease activity as DAS > 1.7 and < 2.6 (13 patients), and high disease activity as DAS28 above 5.1 (9 patients). All controls had CRP within normal limits and did not have any chronic disease with an inflammatory background.
Differences in DReff between RA patients and controls
PBMCs isolated from RA patients were more sensitive to TBH as compared to healthy patients (Figure 1 A; median RA = 13.515; 25% = 10.090; 75% = 13.515 vs. 6.350; 25% = 3.685; 75% = 8.540; Mann-Whitney rank sum test p < 0.001). Moreover, the RA group has more subjects in groups classified as low efficient and inefficient DNA repair as compared with the control (Figure 1 B and Table I). Similarly, we observed more people with inefficient DNA repair in the RA group treated with DMARDS (Table I).
Figure 1
A – Peripheral blood mononuclear cells (PBMCs) show greater sensitivity to tert-butyl hydroperoxide (TBH) at 7 µM for 15 min as scored by DNA damage. PBMCs were isolated from 30 healthy controls (white box) and 30 rheumatoid arthritis (RA) patients (grey box). B – Distribution of individual DNA repair efficiency in 30 healthy controls (white box) and RA patients. Data are presented as median. Differences between groups were analyzed using the Mann-Whitney rank sum test. ***p < 0.001
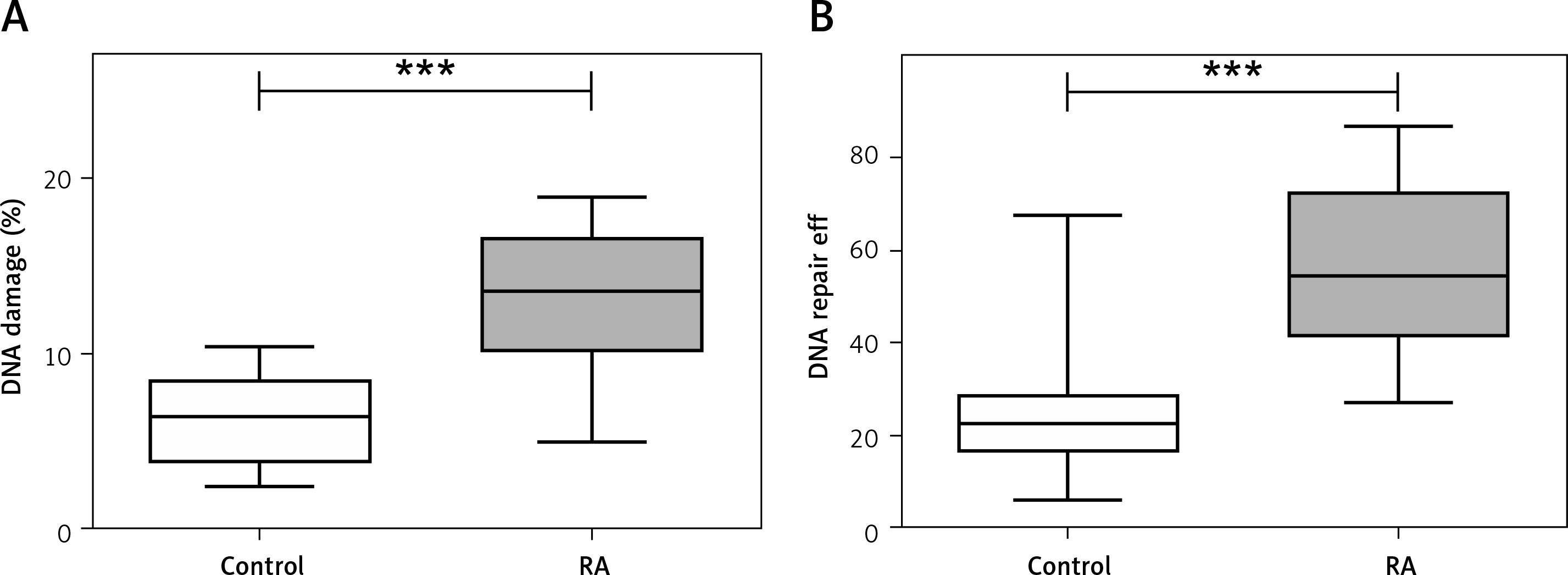
Table I
Efficiency* of repair of DNA lesions induced by TBH (the lower the value, the better)
Differences in BER mRNA expression levels between RA patients and controls
We also observed a generally lower expression level of BER genes in RA patients than controls. A statistically significant difference in the level of gene expression was calculated for: OGG1, MUTYH, NTHL1, LIG3, PARP3, APEX1, APEX2, MBD4 and PARP1 genes (Table II). Next, the relative expression levels were grouped into quartile values of the controls to use in multivariate logistic regression analysis. Notably, in some subjects, expression of UNG, PARP1 and PARP was practically undetectable, and the median was 0.
Table II
Comparison of the mRNA expression levels of 16 core base excision repair genes between 30 rheumatoid arthritis (RA) patients and 30 controls. Analysis was performed using PrimeTime qPCR Assay. All expression values were multiplied by 103 for better presentation of data
| Gene | Median/IQR | P-value* | |
|---|---|---|---|
| RA (n = 30) | Control (n = 30) | ||
| OGG1 | 3.102/5.719 | 6.302/10.883 | 0.006051 |
| OGG1# | 2.925/10.598 | 9.165/11.264 | 0.0003 |
| MUTYH | 0.000/0.730 | 0.796/1.055 | 0.008 |
| MUTYH# | 0.467/1.455 | 0.860/2.991 | 0.0611 |
| NTHL1 | 1.982/4.996 | 3.19/5.824 | 0.0479 |
| NEIL1 | 0.67/1.592 | 0.771/3.022 | 0.3208 |
| NEIL2 | 3.631/12.007 | 7.124/0.139 | 0.1462 |
| UNG | 0.000/1.001 | 0.502/0.868 | 0.4404 |
| TDG | 1.484/9.673 | 1.696/4.013 | 0.7635 |
| LIG3 | 1.919/5.64 | 4.231/5.954 | 0.0076 |
| PARP3 | 0.000/0.001 | 0.226/0.907 | 0.0005 |
| PARP3# | 5.439/10.344 | 7.274/12.384 | 0.0964 |
| MPG | 5.439/10.100 | 7.274/12.384 | 0.0995 |
| APEX1 | 12.082/29.179 | 22.290/15.943 | 0.0274 |
| APEX2 | 20.980/29.314 | 29.038/26.569 | 0.0479 |
| MBD4 | 0.853/1.736 | 2.167/4.465 | 0.0001 |
| NEIL3 | 21.043/48.529 | 26.42/33.452 | 0.5324 |
| PARP1 | 0.000000/0.003974 | 5.031/10.653 | 0.0047 |
| SMUG1 | 7.724/34.175 | 20.019/27.396 | 0.6228 |
Allele and genotype distribution of key BER genes in RA patients and controls
The observed differences in the expression of key genes encoding BER proteins were not due to a change in the profile of CpG island methylation (Supplementary Figure S1).
RA and control groups were similar in the allele and genotype distribution (Supplementary Table SI). No association with RA was found using univariate logistic regression. This is not surprising given the sample size and aims of this study, since we focused on correlation of phenotype with genotype (SNP) and epigenetic factors (expression ratio).
Associations between DReff and RA risk
To estimate RA risk, the relative DReff were grouped into quartile values of the controls (Tables I and III). The crude ORs for RA risk associated with relative DReff in the second quartile, third quartile and fourth quartile were 2.4 (95% CI: 0.34–17), 32 (95% CI: 4.6–222.6) and 104 (95% CI: 8.5–1279.2), compared with the first quartile (the highly efficient DNA repair). After adjusting for SNP rs246079 in UNG gene and expression level of UNG, MUTYH and NEIL3 genes in multivariate logistic regression analysis, the ORs of DReff increased significantly in the third and fourth quartile, corresponding to the low efficient and no repair group, respectively.
Table III
Logistic regression analysis of DNA repair efficiency in RA cases and controls
We also tested potential correlations between DReff and clinical parameters of RA such as DAS, RF, aCCP and CRP, but no correlations were found.
Discussion
We observed an association between RA occurrence, impaired DNA repair of oxidative DNA lesions in PBMCs, polymorphism of the UNG gene and expression of key BER genes – UNG, MUTYH and NEIL3. Therefore, our results suggest that the genetic variations within BER genes as well as epigenetic factors may be linked with RA by the modulation of the cellular response to oxidative stress, and this polymorphism may be a useful additional marker in this disease and/or an environmental indicator of oxidative stress. Because of oxidative stress, mainly 8-oxo-7,8-dihydroguanine (8-oxoGua) and 2,6-diamino-4-hydroxy-5-formamido-pyrimidine were formed in DNA, which arise as a result of attaching to the guanosine ring at the C8 position of the hydroxyl radical and their further transformations in redox reactions [30]. These DNA lesions are the main substrate for the BER pathway and are removed with the help of MUTYH and NEIL3 DGs. MUTYH is responsible for the removal of incorrectly substituted adenine and 2-hydroxyadenine paired with 8-oxo-dG, whereas NEIL3 exhibits a broad substrate recognition spectrum and can excise both oxidized pyrimidines and purines except 8-oxo-G [31]. A few reports have been published describing the effect of altered expression of BER genes in RA. Most of them associated the PARP1 gene with the progression of inflammation in RA (reviewed in [32]). The second concerns the MUTYH gene. The observed serum level of this gene was 8.8% higher in RA than in healthy people [33]. Our research clearly indicates that patients with RA have a reduced expression level of genes encoding two important DGs involved in repair of oxidative DNA lesions in PBMC compared to the control group. Decreased gene expression may result in ineffective or complete lack of repair, and our study is in line with this hypothesis. Unrepaired damage can lead to formation of double-stranded DNA strand breaks (DSBs) as a result of further cellular metabolism [34]. DSBs are among the most serious forms of DNA damage, often resulting in cell death, aging or mutations. The latter can lead to the development of cancer, which accounts for the significant number of RA patients who develop cancer. The mutagenic effects of oxidative DNA damage are well recognized and associated with DSBs. For example, GC → TA and CC → TT are signature mutations for oxidative DNA lesions, and they have been observed in the p53 tumor suppressor or the ras oncogene gene in various cancers.
The finding suggesting the potential involvement of UNG at the genetic and epigenetic levels in the inefficient repair of oxidative lesions in RA is exciting. UNG is the primary glycosylase involved in the removal of uracil (U) from DNA. The involvement of UNG in the removal of oxidative DNA lesions is minimal in contrast to other glycosidases; however, it plays a very important role in promoting lymphocyte development. During lymphocyte development, two molecular processes in DNA occurred – V(D)J recombination and class-switch recombination (CSR). Both are important for the formation of a wide range of lymphocyte B antigen receptors and heavily depend on somatic hypermutation (SHM). SHM is conducted in cooperation with BER and MMR systems and UNG plays a key role in this process by recognizing and processing U. Imbalance of UNG level increased the risk of developing B-cell lymphomas. However, it is not only carcinogenesis that should be addressed, as UNG is also likely involved in development of autoimmune diseases. The involvement of UNG in autoimmune diseases is complex, and loss of UNG or changes of its activity via SNP can affect RA progression in two ways. Firstly, it can be responsible for severely impaired CSR and antibody affinity maturation via SHM [35]. Secondly, it can induce apoptosis in B cells. Apoptosis results in release of DNA fragments outside the cell. DNA fragments can stimulate innate inflammatory responses via activation of the stimulator of interferon genes adaptor protein (reviewed in [36]). This can produce a feedback loop, as inflammation induces production of ROS that in turn generate oxidized DNA lesions, which – if not repaired – lead to apoptosis.
These findings confirm those of our earlier research [37], and also suggest that SNPs in DNA repair genes may be additional genetic factors associated with RA and contribute to the etiology, pathogenesis and outcome of RA. However, the groups were too small to enable a final conclusion, and further studies are required to address the question.
Although our study has the character of basic research, clinical aspects can be derived from it. The antioxidant potency of the host should be increased in order to avoid the effects of oxidative stress at the DNA level in people with reduced DNA repair efficiency. This can be done with an antioxidant-rich dietary intervention, because such a diet ameliorates the state of oxidative stress and improves the antioxidant potency [38, 39]. In addition, we believe that a closer understanding of the genetic background of RA in association with the clinical phenotype will allow for future personalized treatment of RA.
Our study has some limitations. First of all, the influence of the drugs taken by the patient cannot be neglected. MTX and GCS have a plethora of effects on RA patients. MTX but not GCS was associated with a slightly higher level of endogenous DNA damage compared with RA patients without treatment, as we reported previously [21]. However, considering the research hypothesis of this article, it is irrelevant whether the observed alterations in the activity of DNA repair processes and their association with genotype or gene expression alterations are due to RA phenomena or therapy. Nonetheless, we are more convinced of the former option, since MTX and GCS are drugs which are generally considered safe, and no association between treatment with these drugs and cancer development has been demonstrated. Regardless, our study had mainly a preliminary character, and these results need confirmation in larger cohorts outside the Central European populations, as the main limitations of the study are its single-center design, sample size and ethnicity.
In conclusion, impaired repair of oxidized DNA lesions in RA patients may accelerate PBMC aging and contribute to the RA phenotype and association of RA with cancer development. Genetic and epigenetic factors contribute to this phenomenon, but epigenetic factors that lead to lower expression of BER genes need to be identified. We excluded the possibility of a different methylation pattern in RA patients as an epigenetic factor. Thus, we must consider the two latter options: histone code and miRNA expression. The last one is particularly interesting, as some miRNAs are being considered as biomarkers for RA, but they need validation in a real-world clinical setting (reviewed in [40]). A potential link between RA and ineffective repair of oxidative DNA damage may be miRNA-155, as it is associated with the occurrence of both phenomena [41, 42].