A 73-year-old man was referred to our hospital with intermittent diarrhea and a generalized scattered red rash for 5 days. He had a previous history of gallbladder stones and had no episodes of biliary colic; no significant family history was present. On examination, he had no fever and presented with a generalized scattered patchy red rash with pruritus, and several enlarged lymph nodes that were hard, nontender, movable, and approximately 5 mm in diameter could be palpated in the bilateral axillae. Routine blood tests on the first day of admission were normal, with only the lactate dehydrogenase elevated to 498 IU/l (normal range: 120–250). He was subsequently given dexamethasone and cimetidine to treat the rash. Over the next 1 week, he developed recurrent hyperthermia without any infectious conditions, with fever peaking over 38.5°C. The second laboratory test showed a white blood cell count of 0.34 × 109/l (normal range: 4–10), neutrophil count of 0.01 × 109/l (normal range: 2–7), neutrophil percentage of 10.7% (normal range: 50–70), hemoglobin of 96 g/l (normal range: 110–150), platelets of 35 × 109/l (normal range: 100–300), high-sensitivity C-reactive protein greater than 10 mg/l, fibrinogen of 1.44 g/l (normal range: 2–4), D-dimer level of 1.68 µg/ml (normal range: 0–0.5), lactate dehydrogenase of 311 IU/l, serum ferritin level of 599.7 ng/ml (normal range: 15–200) and triglycerides of 3.1 mmol/l (normal range: 0–1.7). NK cell activity was reduced or even non-existent. Contrast-enhanced CT of the abdomen revealed multiple enlarged lymph nodes in the hepatogastric space and retroperitoneum. Cranial and extremity bone radiographs indicated osteoporosis with no abnormal bone destruction. He was first considered for lymphoma, but biopsies of bilateral axillary lymph nodes revealed diffuse hyperplasia of Langerhans histiocytes, and immunohistochemistry showed S100 (++) and CD1a (++). Based on these findings, he was considered for a diagnosis of Langerhans cell histiocytosis (LCH) with a multisite type. Subsequently, bone marrow puncture clearly revealed hemophagocytosis. Epstein-Barr virus and cytomegalovirus DNA tests were negative. There were no gene mutations seen in the genetic testing associated with hemophagocytic lymphohistiocytosis (HLH). The final diagnosis of LCH combined with secondary HLH was made according to HLH-2004 criteria (not meeting molecular biology diagnostic criteria but meeting at least five of the eight clinical and laboratory indicators), after excluding leukemia and lymphoma [1]. Desloratadine and promethazine were administered to treat the rash, recombinant human granulocyte colony-stimulating factor (rhG-CSF) to raise leukocytes, dexamethasone to attenuate the inflammatory storm, and itraconazole to prevent fungal infection. In the third week of hospitalization, the patient’s symptoms were significantly better than before, with a rechecked leukocyte count of 15.2 × 109/l, neutrophil count of 9.21 × 109/l, hemoglobin of 101 g/l, and platelets of 93 × 109/l, but he finally refused chemotherapy and was discharged. The patient died of severe lung infection and uncontrollable HLH 1 month later.
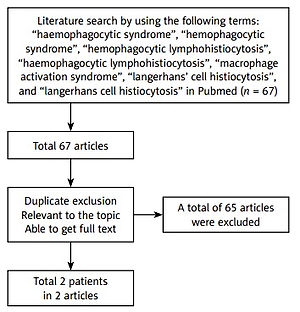
We conducted a systematic literature review of publications in PubMed up to and including April 1, 2022. The following terms were used: “haemophagocytic syndrome”, “hemophagocytic syndrome”, “hemophagocytic lymphohistiocytosis”, “haemophagocytic lymphohistiocytosis”, “macrophage activation syndrome”, “langerhans’ cell histiocytosis”, and “langerhans cell histiocytosis”. We included only adult patients (age ≥ 18) with a diagnosis of LCH and HLH mentioned in the literature; articles containing terms of suspected diagnosis and possible diagnosis and those included in studies of juveniles (age < 18) were excluded (Figure 1). The literature search yielded 2 eligible articles after excluding duplicate publications, unrelated to the topic, and unavailable full-text articles [2, 3]. A total of 3 patients were identified, with a female/male ratio of 1/2, a mean age of 75.6 years (range: 73–78), and a median age of 76 years based on the available data (Table I).
Table I
Clinical features of three adult patients with LCH combined with secondary HLH
| Authors | Sex/age [years] | Symptoms | Medical history | Tissue biopsy | Treatment | Prognosis |
|---|---|---|---|---|---|---|
| Chamorro et al. [2] | Female/76 | Febrile and cachectic, with firm, red-brown papules on the torso | / | Histiocytes within the papillary dermis staining S100 and CD1a positive | / | / |
| Ioannidou et al. [3] | Male/78 | Rapidly spreading psoriasiform eruption; fatigue; cough; generalized weakness and fever | Smoke | Skin biopsy showed infiltration of the papillary dermis with a dense cellular infiltrate. Immunopositivity with antibodies against CD1a, S100 protein and HLA-DR was positive | Ceftazidime; netilmicin; etoposide; prednisolone | Died of pulmonary abscess and sepsis |
| This case | Male/73 | Intermittent diarrhea, generalized scattered red rash, and fever | Gallbladder stones | Biopsy of bilateral axillary lymph nodes revealed diffuse hyperplasia of Langerhans histiocytes, and immunohistochemistry showed S100 (++) and CD1a (++) | Desloratadine; promethazine; rhG-CSF; dexamethasone; itraconazole | Died of severe lung infection |
LCH is a disease mostly seen in children in which LCH cells derived from myeloid dendritic cells proliferate and invade multiple or single systems [4]. LCH is rare in adults, and much of what we know about its treatment and diagnosis comes from pediatric studies [5, 6]. HLH represents a spectrum of hyperinflammatory syndromes typically characterized by dysregulated activation of the adaptive and innate immune systems [7, 8]. LCH and HLH are both histiocyte-derived diseases [8]. Tang et al. summarized 57 patients with LCH-associated HLH and reported that the median age of LCH onset was 1 year, and 91% (52/57) of patients diagnosed with LCH were less than 2 years old. Ninety percent of LCH patients developed HLH at the diagnosis or during chemotherapy. Of the 57 LCH-associated HLH patients, 15 died [9]. The coexistence of these two diseases in adults is of particular interest, as to date the vast majority (almost all) of cases occur in children and only 2 cases have been reported in the adult population. From the tabulated data, fever and skin lesions with systemic multisystem involvement were common in all patients, and lymph node and bone marrow biopsies were essential for a definitive diagnosis. However, the majority of patients had a poor prognosis, as demonstrated by the deaths of 2 patients from pulmonary infection, which was closely related to the poor resistance of the elderly and to the immune compromise caused by the disease itself, which led to secondary pulmonary infection. Therefore, the importance of early prophylactic anti-infection is emphasized. Definitive diagnosis of these two diseases in the adult population may be challenging. We should look for macrophage activation in patients with evidence of hyperacute clinical deterioration or multisystem LCH. In addition, the patient in this paper had a rash and diarrhea as the first manifestations. LCH in adults is often restricted to the skin before becoming systemic and can mimic other common skin diseases such as seborrhoeic dermatitis, psoriasiform lesions, erythema, pustules, rashes, and erosions. It is difficult to make a diagnosis based on cutaneous manifestations alone, thus emphasizing the importance of skin biopsy. Previous studies have shown that LCH itself can cause gastrointestinal symptoms [10]. However, LCH with gastrointestinal involvement is rare, with a poor prognosis, and an endoscopic and skin biopsy might have been beneficial for early diagnosis in our patient.
In conclusion, on the one hand, LCH has an insidious onset, and on the other hand, patients with LCH who have fever and skin lesions with systemic multisystem involvement need to be particularly alert to the possibility of combined HLH. In patients with secondary HLH, the cause should be actively investigated to improve survival.