Introduction
Lung cancer is the leading cause of cancer incidence and mortality worldwide [1]. Non-small cell lung cancer (NSCLC), which accounts for about 80% of all lung cancers, has a low 5-year survival rate [2]. Furthermore, about 75% of NSCLC patients are already in the advanced stage when they are properly diagnosed with NSCLC [3]. In addition, the effects of the clinical NSCLC treatments are still unsatisfactory. Patients who have undergone surgical resection have high recurrence rates. On the other hand, chemotherapy or radiotherapy always causes many side effects. Thus, more effective diagnoses and therapeutic options are urgently needed for patients who suffer from NSCLC.
MicroRNAs (miRNAs) are a type of highly conserved endogenous non-coding fractions of approximately 18 to 25 nt RNAs that play important roles in regulating cell differentiation, apoptosis, tumorigenesis and other biological processes by targeting mRNAs for cleavage or translational repression [4–8].
In the past few years, miRNAs have been found to be closely related to lung tumor biology [9, 10]. For example, Let-7 serves as an anti-tumor microRNA in lung cancer through its ability to downregulate the expression of RAS, which plays oncogenic roles in many kinds of tumors. Reduced expression of Let-7 is observed in lung cancer and is associated with shortened postoperative survival [11, 12]. MiR-340 suppresses lung cancer proliferation and induces apoptosis by enhancing p27 signaling, which can inhibit the activity of cell cycle protein cyclin-dependent kinase 2 (CDK2) [13]. Therefore, elucidating the underlying biological functions and molecular mechanisms of miRNAs in lung cancer will benefit our understanding of miRNAs in lung cancer therapy.
MiR-1286 is a newly identified microRNA located on human chromosome 22. It was reported that the expression of miR-1286 changed during brain development [14]. In another study, miR-1286 was found to be hypermethylated in breast cancer tumors compared with normal tissues, suggesting a potential role of miR-1286 in modulating cancer development [15]. However, as far as we know, the exact function of miR-1286 and its mechanism of action in cancer have never been reported and the potential targets of miR-1286 in lung cancer have not yet been identified.
Tumors need more nutrients and energy to sustain their rapid proliferation, thus resulting in abnormal glucose, lipid and protein metabolism. For example, even in the presence of sufficient oxygen, tumor cells tend to favor metabolism through glycolysis rather than the oxidative phosphorylation pathway, simultaneously accompanied by high glucose utilization and production of large amounts of lactate [16]. The above phenomenon is known as the Warburg effect. Pyruvate kinase muscle isozyme (PKM), including PKM1/M2, catalyzes the last step in the course of aerobic glycolysis [17]. A recent publication reported that miR-124, miR-137, and miR-340 impair colorectal cancer growth by counteracting the Warburg effect due to interference with PKM [18]. In addition, miR-148a/152 suppresses the expression of PKM2, which can increase the activity of pro-proliferating NF-κB signaling, by which miR-148a/152 inhibits the growth and angiogenesis of cancer cells [19]. However, whether miR-1286 is involved in regulating aerobic glycolysis in cancer cells remains unknown.
Here we demonstrate for the first time that miR-1286 is down-regulated in lung tumor tissues compared with adjacent normal tissues. Then, miR-1286 inhibits lung cancer cell growth by decreasing PKM2 expression. A549 cells with miR-1286 overexpression have a significantly lower growth rate in vitro and in vivo. Thus, our study reveals the crucial role of miR-1286 in lung cancer and may have important clinical implications in lung cancer therapy.
Material and methods
Human samples
NSCLC tissues and paired adjacent noncancerous tissues were obtained from patients diagnosed with NSCLC in the Second Affiliated Hospital of Xingtai Medical College Hospital. Total RNA was extracted using TRIzol according to the standard procedure and prepared for quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analysis. Studies involved in clinical specimens were approved by the Ethics Committee of the Second Affiliated Hospital of Xingtai Medical College Hospital.
Reagents
Anti-PKM2 antibody (D78A4, 1 : 1000), anti-β-actin antibody (8H10D10, 1 : 3000), HRP-conjugated polyclonal goat anti-mouse antibody (#7076, 1 : 2000) and HRP-conjugated polyclonal goat anti-rabbit antibody (#7074, 1 : 2000) were from Cell Signaling (Danvers, MA, USA). The lactate assay kit (MAK64) was purchased from Sigma-Aldrich (USA). Cell Counting Kit-8 (CCK8) was bought from Transgen Biotech (Beijing, China).
Cell transfection
Human NSCLC cell line A549 cells were purchased from American Type Culture Collection (ATCC). Cells were cultured in F-12K culture medium (Thermo Fisher Scientific, USA) containing 10% fetal bovine serum (Gibco, USA) at 37°C with 5% CO2. The miR-1286 mimics, miR-1286 inhibitors and negative control sequences were all purchased from GenePharma (Shanghai, China). Cells were divided into four groups: miR-1286 mimic group vs. control mimic group, miR-1286 inhibitor group vs. control inhibitor group. 1 × 105 A549 cells were plated into 24 wells overnight. When the cell density reached 50–60%, A549 was transfected using TransIT transfection reagent (Mirus) according to the manufacturer’s instructions. Forty-eight hours after transfection, cells were collected for detection. For A549 subcutaneous inoculation, A549 cells were transfected with pLenti-III-miR-1286 lentivirus or control lentivirus, and selected by puromycin for 5 weeks to acquire cells that stably overexpress miR-1286.
Cell proliferation assay
After being transfected by miR-1286 mimic or miR-1286 inhibitor for 48 h, A549 cells were separately seeded in 96-well plates at a density of 3 × 103 cells/well in 100 μl medium and five duplicates for each group. The rate of cell proliferation was detected at the time points of 18 h and 36 h. 10 μl of CCK8 were added to each well and the cells were incubated for an additional 4 h. The absorbance value at 450 nm was then recorded and calculated. Meanwhile, cells were collected and counted by the plate counting method to determine cell number at 36 h.
RNA extraction and q-PCR
Total RNA was extracted with Trizol (Invitrogen, USA) and cDNA was synthesized using a PrimeScript RT reagent kit (Takara, Japan) according to the manufacturer’s instructions. Quantitative RT-PCR (qRT-PCR) was performed in an Applied Biosystems 7500 real-time PCR system (Foster City, CA, USA) through the application of SYBR Green PCR master mix (TaKaRa Biotechnology Co. Ltd). All quantitative RT-PCR reactions were performed in triplicate. The relative mRNA levels of miR-1286 and PKM2 were normalized against β-actin RNA. The corresponding PCR primers were as follows: PKM2 forward 5′-ATGTCGAAGCCCCATAGTGAA-3′ and reverse 5′-TGGGTGGTGAATCAATGTCCA-3′; β-actin was used as an internal control and was amplified using primers 5′-AGTGTGACGTTGACATCCGT-3′ and 5′- GCAGCTCAGTAACAGTCCGC-3′.
Western blotting
Total protein of A549 cells was isolated with cell lysis buffer (Cell Signaling Technology) and quantified using the BCA protein assay. 20 μg of total proteins were separated by 10% SDS-PAGE gel and transferred onto a PVDF membrane (Millipore, Billerica, MA). The corresponding primary antibodies were incubated overnight. The corresponding HRP-conjugated secondary antibody was then added and incubated with membranes for 1.5 h at room temperature. Protein bands in PVDF membranes were visualized using SuperSignal West Femto Maximum (Thermo, IL, USA) and detected by BioImaging Systems (Tanon). All samples were assayed in triplicate.
Lactate production assay
A549 cells post-treated by miR-1286 mimic or miR-1286 inhibitor were seeded in 6-well plates in fresh medium for 24 h. Then, detection of lactate was performed using a lactate assay kit. A549 supernatants were collected by centrifugation at the speed of 600 g for 5 min. Lactate concentration in A549 supernatants was measured using a Lactate Assay Kit (Sigma Aldrich, USA) according to the manufacturer’s instructions on a Nova Bioprofile 100 analyzer. (Nova Biomedical, USA)
In vivo tumor xenograft assay
Six-week-old female athymic nude mice were purchased from SIPPR-BK Experimental Animal Co. (Shanghai, China) and kept in a specific pathogen-free facility. A549 cells infected with lentivirus expressing miR-1286 or control were suspended in PBS at the density of 2 × 107 cells/ml. Subsequently, a 100 μl cell suspension was injected subcutaneously into the flank region of the nude mice. Every 4 days, tumor size was measured by a caliper and tumor volume was calculated by length × width2/2. Three weeks later, mice were sacrificed and tumor tissues were collected for further analysis.
Statistical analysis
All data represent at least three independent experiments to assess statistical significance. Data are shown as the mean ± SD. P < 0.05 was considered as statistically significant and indicated by *p < 0.05, **p < 0.01, ***p < 0.001. All data analysis was performed with SPSS 16.0 software (SPSS Inc., Chicago, IL) and analyses used in this study mainly included two-way ANOVA, paired t test and Student’s t test.
Results
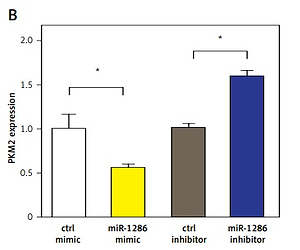
MiR-1286 is down-regulated in NSCLC tissues
In order to investigate the potential role of miR-1286 in NSCLC, we firstly collected 14 pairs of NSCLC tissues and matched noncancerous adjacent tissues, and performed q-PCR to evaluate miR-1286 expression. As shown in Figure 1, miR-1286 expression levels in NSCLC tissues were significantly lower than in corresponding noncancerous adjacent tissues, hinting that miR-1286 may take part in the tumorigenesis process of NSCLC.
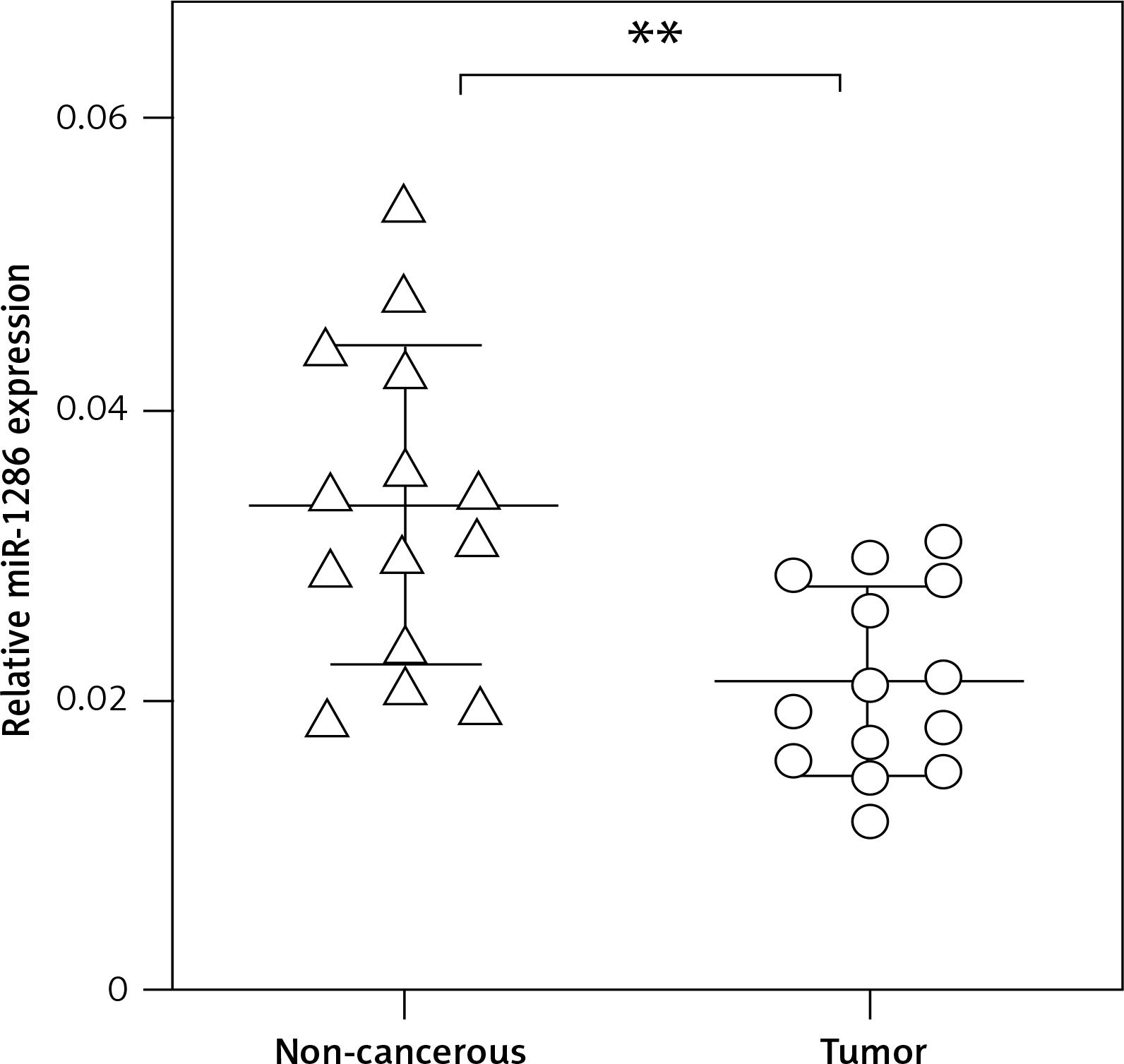
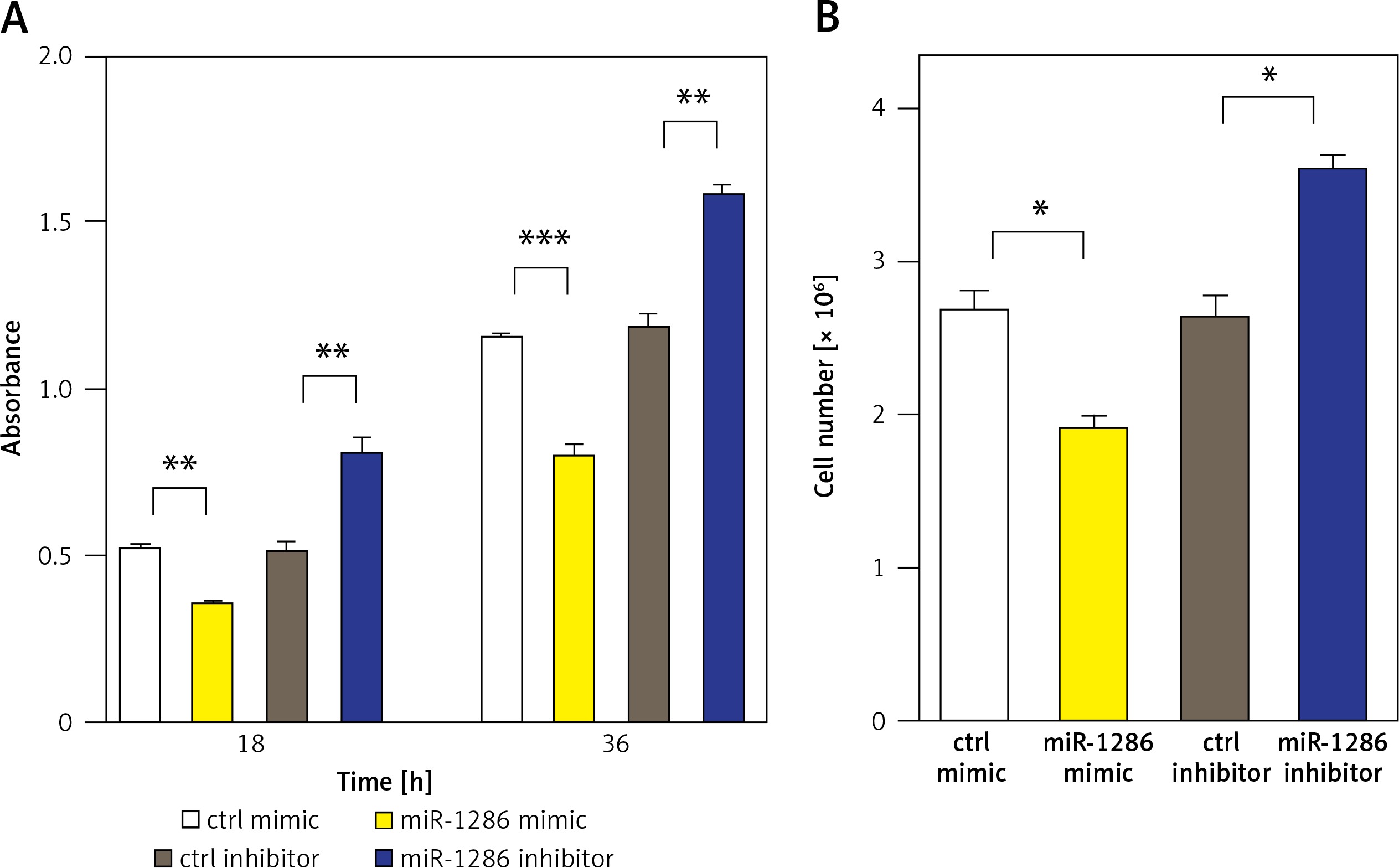
MiR-1286 strongly inhibits NSCLC cell proliferation in vitro
We next evaluated the effect of miR-1286 on the proliferation of NSCLC cells. MiR-1286 overexpression in A549 cells was conducted by transfecting miR-1286 mimic or control mimic. Subsequently, CCK8 assay was performed to detect the level of cell proliferation. The results showed that compared with control mimic, miR-1286 mimic decreased A549 cell proliferation. In contrast, A549 cells treated with miR-1286 inhibitor exhibited a significant increase in growth (Figure 2 A). The suppressive role of miR-1286 on A549 cell proliferation was further verified by cell number counting (Figure 2 B). Taken together, these results suggest that miR-1286 inhibits the proliferation of NSCLC cells in vitro.
Figure 2
MiR-1286 overexpression inhibited the proliferation of NSCLC cells. A – CCK8 assay was performed to detect A549 cell proliferation ability at indicated time points after transfection of miR-1286 mimic or control mimic and miR-1286 inhibitor or control inhibitor. B – Cell number was determined by cell counting after transfection of miR-1286 mimic or control mimic and miR-1286 inhibitor or control inhibitor for 48 h in A549 cells
*P < 0.05, **p < 0.01.
PKM2 is the target of miR-1286
MiRNAs usually exert their functions by binding to the 3′-UTR of their direct target genes thus downregulating their expression [20–23]. To identify potential downstream targets of miR-1286, we checked validated targets in the Probability of Interaction by Target Accessibility (PITA) database. Interestingly, we found that PKM2 was the target gene of miR-1286 (Figure 3 A). The PKM2 expression was decreased in miR-1286-overexpressing A549 cells, while PKM2 expression was increased in miR-1286-inhibitor treated A549 cells, which were verified by both q-PCR and western blot analysis (Figures 3 B, C).
Figure 3
PKM2 was a target gene regulated by miR-1286. A – PTIA database was used to scan the matched sequence, and then PKM2 gene was predicted as a direct target of miR-1286. B, C – A549 cells were either transfected with miR-1286 mimic and control mimic or treated with miR-1286 inhibitor or control inhibitor respectively. The mRNA expression of PKM2 was analyzed by qRT-PCR after transfection for 24 h (B) and the protein level of PKM2 was measured by western blotting after transfection for 36 h (C). D – After A549 cells were either transfected with miR-1286 mimic and control mimic or treated with miR-1286 inhibitor or control inhibitor for 24 h, A549 supernatants were collected to detect the production of lactate. E – The expression of several glycolysis-related enzymes was evaluated by qRT-PCR, including hexokinase 1 (HK1), hexokinase 2 (HK2), phosphofructokinase 1 (PFK1) and lactate dehydrogenase (LDH). F – The correlation between expression of PKM2 and miR-1286 in A549 cells was analyzed by Spearman correlation analysis
*P < 0.05, **p < 0.01.
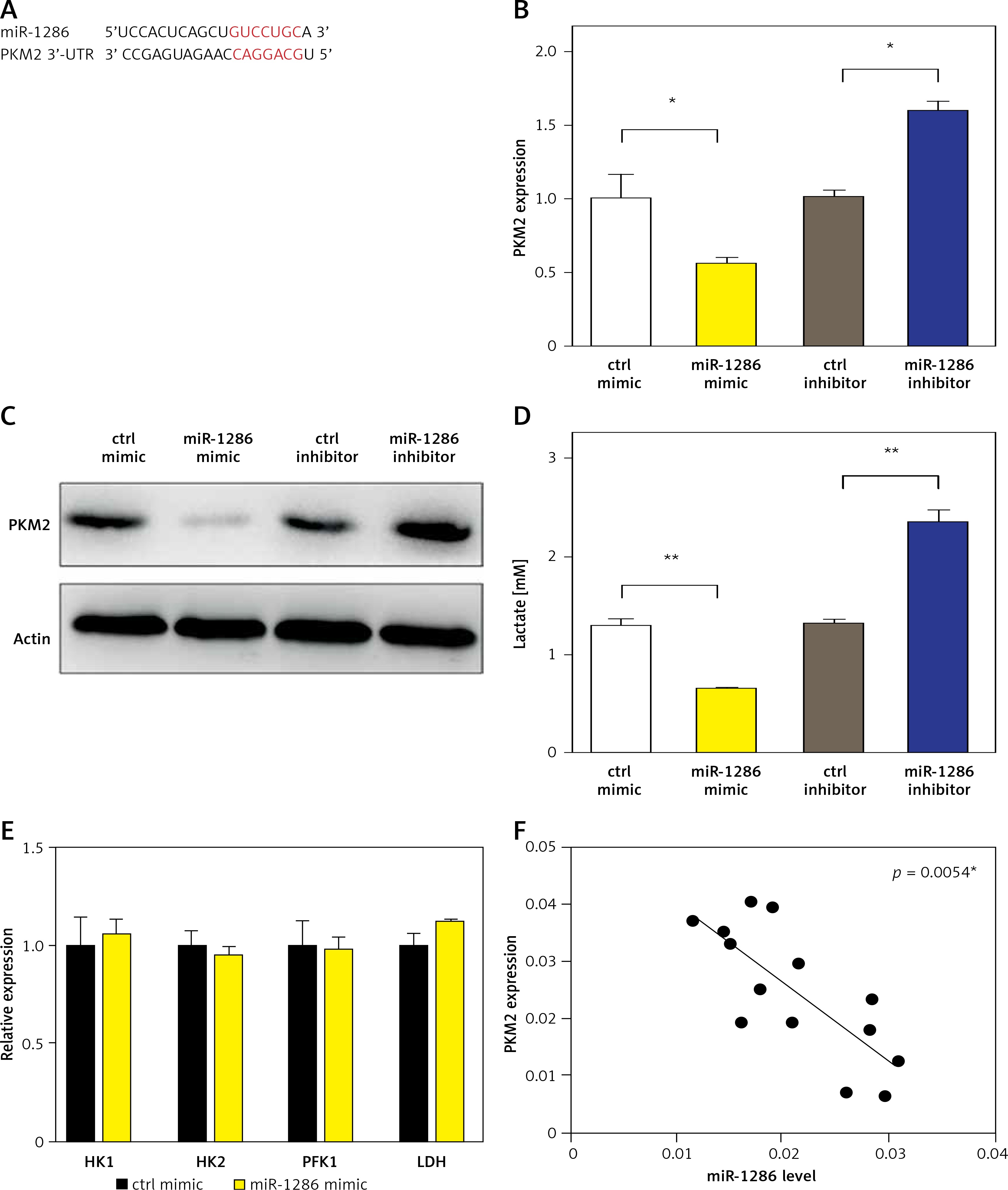
Cell metabolism reprogramming, especially altered glucose metabolism, is vitally essential for tumorigenesis. Tumor cells preferentially use aerobic glycolysis to generate energy and support their fast proliferation. Particularly, PKM2 is known to be a crucial regulator in the glycolytic pathway [24, 25]. Therefore, we detected the level of lactate, the final product of glycolysis, in A549 culture supernatants. In comparison with the control group, miR-1286 overexpression led to less lactate production, while miR-1286-inhibitor treated A549 cells produced more lactate (Figure 3 D). On the other hand, the expression of several other glycolysis-related enzymes, including hexokinase 1 (HK1), hexokinase 2 (HK2), phosphofructokinase 1 (PFK1) and lactate dehydrogenase (LDH), showed no significant difference between miR-1286 mimic and control mimic cells (Figure 3 E). To investigate the role of miR-1286 in glycolysis, we analyzed the correlation of miR-1286 and PKM2 and found that the expression level of PKM2 was negatively correlated with miR-1286 (Figure 3 F). These data suggest that PKM2 is the target gene of miR-1286 and miR-1286 may take part in regulating the aerobic glycolytic pathway.
MiR-1286 modulates NSCLC cell proliferation via targeting PKM2
In order to clarify whether the inhibitory effect of miR-1286 on lung cancer cell proliferation was dependent on PKM2 downregulation, A549 cells were co-transfected with miR-1286 mimic and PKM2. As shown in Figure 4 A, in contrast with the control group, PKM2 overexpression partially reversed the ability of miR-1286 mimic to repress cell proliferation. Correspondingly, inhibition of miR-1286 expression accelerated cell proliferation, which would be abolished by knockdown of PKM2 expression (Figure 4 B).
Figure 4
MiR-1286 inhibited NSCLC cell proliferation via targeting PKM2. A – Cell proliferation was determined by CCK8 assay after transfection of miR-1286 mimic or control mimic, together with pcDNA3.1-PKM2 or pcDNA3.1 for 48 h. B – Cell proliferation was determined by CCK8 assay after transfection of miR-1286 inhibitor or control inhibitor, together with shPKM2 or sh-Scramble for 48 h. C – Lactate production was measured by using the Nova Bioprofile 100 analyzer after transfection miR-1286 mimic or control mimic, together with pcDNA3.1-PKM2 or pcDNA3.1 for 24 h. D – Lactate production was measured after transfection miR-1286 inhibitor or control inhibitor, together with shPKM2 or sh-Scramble for 48 h
*P < 0.05, **p < 0.01, ***p < 0.001.
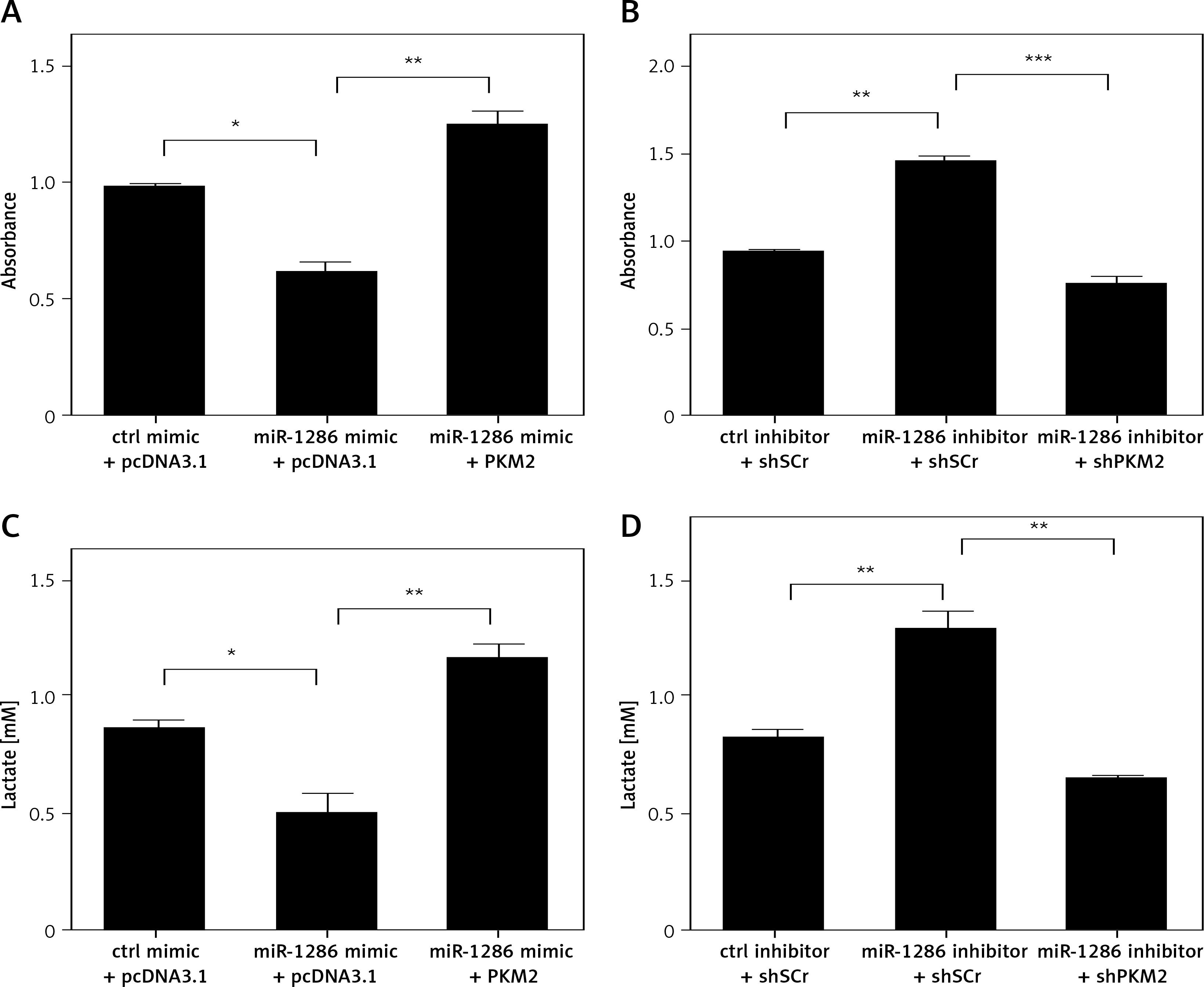
As we previously found that the lactate production could be modulated by miR-1286, it was necessary to confirm whether this regulation function leans on PKM2. A549 cells with high expression of miR-1286 exhibited a decline in lactate production and this effect was inverted by co-transfecting with pcDNA3.1-PKM2 plasmid (Figure 4 C). Meanwhile, A549 cells treated with miR-1286 inhibitor produced less lactate, while decreasing PKM2 expression led to opposite results (Figure 4 D). Taken together, all these results indicate that miR-1286 is likely to participate in the glycolysis process by targeting PKM2 to inhibit NSCLC cell proliferation.
MiR-1286 restrains NSCLC tumor growth in vivo
To determine the role of miR-1286 in tumor development in vivo, we constructed the A549-miR-1286 mimic cell line which could stably overexpress miR-1286. Cells were implanted into nude mice by subcutaneous injection. As shown in Figure 5 A, the tumor volume of the miR-1286 mimic group was significantly smaller than that of the control group. Meanwhile, the tumor tissues were found with higher expression of miR-1286 and a lower level of PKM2 (Figures 5 B, C). The correlation analysis revealed a negative correlation between PKM2 and miR-1286 expression (Figure 5 D).
Figure 5
MiR-1286 affected NSCLC development in vivo. A – Nude mice were subcutaneously injected with 2 × 106 A549-miR-1286 or A549-control cells. Tumor size was monitored over 3 weeks and the tumor volume was calculated. B, C – Expression of miR-1286 (B) and PKM2 (C) in the tumor tissues of A549-miR-1286 or A549-control mice xenograft was detected by q-PCR. D, E – Correlations between expression of PKM2 and miR-1286 (D) or PKM2 and tumor volume (E) in mice tumor tissues were analyzed by Spearman correlation analysis using GraphPad Prism 7
*P < 0.05, **p < 0.01.
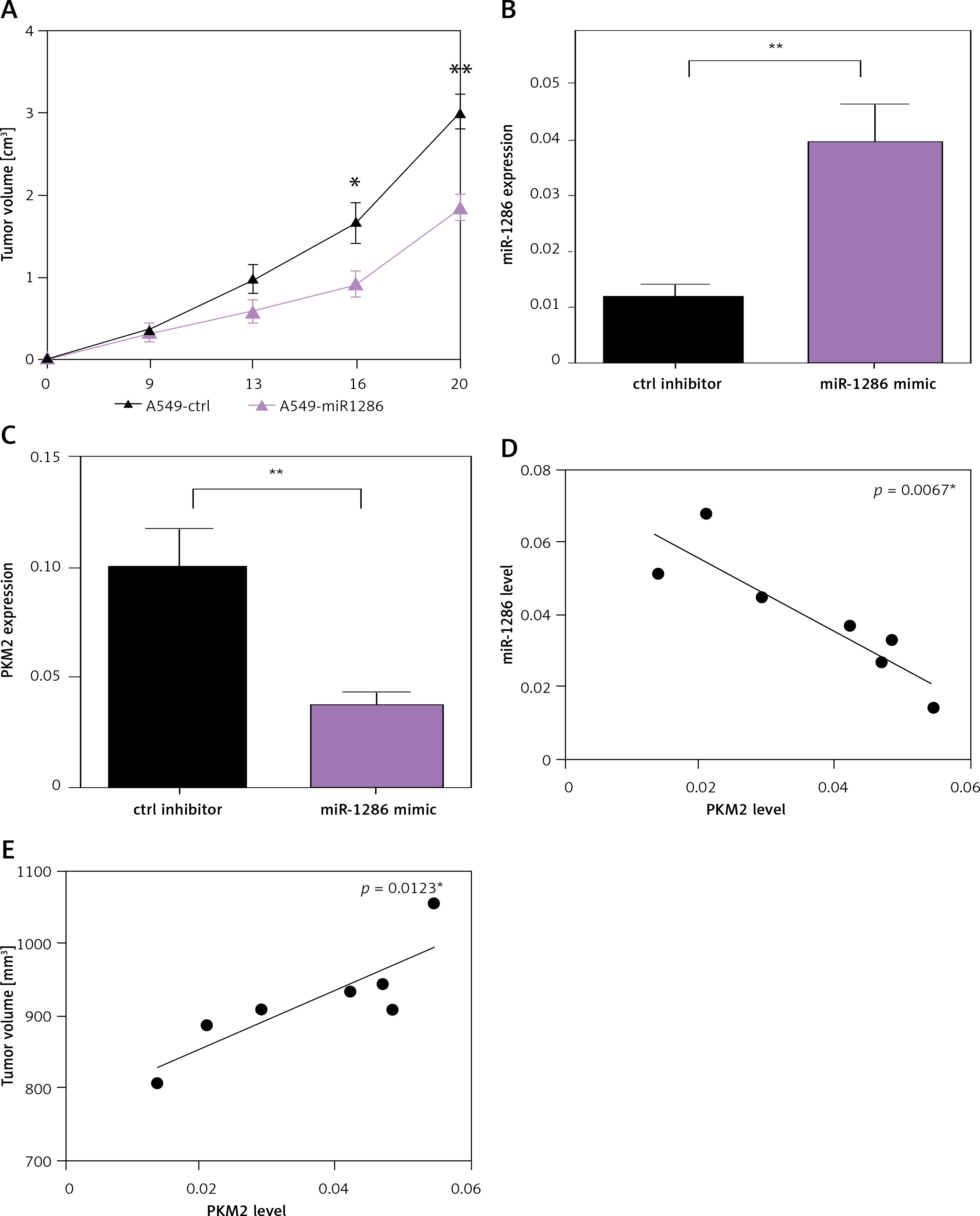
PKM2 has been reported to be closely linked to embryogenesis, tissue repair, and cancer. Studies have found that in regional lymph node metastases in NSCLC patients, there is lower PKM2 expression and better overall survival rate of patients [26]. Hence, we also analyzed the association of PKM2 expression with tumor size in the mouse tumor xenograft model. We achieved a parallel result that the expression of PKM2 was positively correlated with tumor size (Figure 5 E). In sum, these findings suggest that miR-1286 is a key tumor suppressor that restrains NSCLC tumor cell growth in vivo through depending on PKM2.
Discussion
Emerging evidence has shown that abnormal miRNA expression boosts the initiation and progression of various human tumors [27, 28]. A large number of studies have also indicated that miRNAs can be used as target genes for tumor prediction and treatment, including NSCLC [29–31]. For instance, miR-224/-520c decreases the expression of tumor suppressor candidate 3 (TUSC3) and promotes NSCLC metastasis through impairing the unfolded protein response [32]. Administration of nanoparticles containing miR-125b promotes the differentiation of anti-tumor macrophages, thus restraining the growth of NSCLC in vivo [33]. Inhibition of miR-582-3p reduces the malignancy of lung cancer stem cells by suppressing the Wnt/β-catenin pathway [34]. Thus, exploring the role of miRNA in tumors is of great significance for tumor therapy. In our study, we found that miR-1286 was down-regulated in tumor tissues from NSCLC compared with normal tissues for the first time. We also confirmed the negative correlation between miR-1286 expression and A549 cell proliferation in vitro. With regard to the mechanism, functional assays indicated that PKM2 functioned as a downstream target of miR-1286 in NSCLC. Lastly, in vivo experiments again confirmed that restoration of miR-1286 expression in A549 cells impaired their growth in mice. These findings suggest that miR-1286 may serve as a potential anti-tumor microRNA due to its capacity in interfering glycolytic metabolism in tumor cells.
To date, the function of miR-1286 has been poorly investigated. Previous studies only found the altered expression of miR-1286 during brain development and the hypermethylated miR-1286 in breast cancer tissues [14, 15]. To our knowledge, our present work for the first time investigated the regulatory role of miR-1286 in NSCLC. On the other hand, whether miR-1286 can exert a similar function in other types of tumors needs further exploration.
Tumor cells are inclined to favor aerobic glycolysis for glucose metabolism. Previous studies also demonstrated that glucose metabolism modulated by microRNAs is crucial for regulating tumor initiation and progression [35, 36]. There are several rate-limiting enzymes that catalyze the irreversible steps of glycolytic metabolism [37]. When we evaluated the expression of enzymes, including HK1/2, PFK1, LDH and PKM2, only PKM2 expression was found to be downregulated by miR-1286 mimic. Moreover, miR-1286-induced downregulation of PKM2 resulted in the reduced production of lactate, the final product of glycolysis. Therefore, miR-1286 decreases lung cancer cell proliferation via interfering with the Warburg effect by specifically targeting PKM2.
PKM2, which is the terminal glycolytic enzyme pyruvate kinase, is overexpressed in cancer cells [38]. In the past years, studies have focused on the relationship between PKM2 and miRNA, and have revealed that miRNA may have direct or indirect associations with PKM2. Overexpression of miR-122 in human hepatocellular carcinoma significantly reduces the level of PKM2, which increases cell apoptosis and reduces cell migration and invasion [39]. Another report showed that miR-124 functions as a tumor suppressor and modulated energy metabolism through PTB1/PKM1/PKM2 signaling in human colorectal tumor cells [40]. However, in this study, we demonstrated that PKM2 was the target gene of miR-1286 in A549 cells. In addition, miR-1286 was negatively related with PKM2 both in vitro and in vivo. These findings indicate that PKM2 may be investigated as an effective therapeutic target for NSCLC.
In conclusion, our study for the first time demonstrated that PKM2 acts as the target of miR-1286 in NSCLC. Moreover, miR-1286 is lower in tissue tumor from NSCLC compared with normal tissue. Our results contribute to better understanding of miR-1286-mediating lung cancer cell proliferation, which regulates tumor angiogenesis and cancer progression and may provide a novel biomarker and potential therapeutic interventions in the future.