Introduction
Lung cancer (LC) is one of the most common cancers worldwide, and high mortality rates are reported in both males and females for this cancer. It is commonly divided into small cell lung cancer (SCC) and non-small cell lung cancer (NSCLC). Non-small cell lung cancer accounts for the majority of all LCs – more than 80% (it includes adenocarcinomas, large cell carcinomas and squamous cell carcinomas) [1, 2]. Smoking and occupational pollution with substances such as asbestos, lead, copper, arsenic, and polycyclic aromatic hydrocarbons are the most important risk factors for LC. Therefore, early preventive measures and research on the environmental causes of LC are of crucial importance today. Other risk factors for LC include genetic factors, dietary factors, and lung diseases such as pulmonary fibrosis and chronic obstructive lung disease [1]. Moreover, genetic factors are another predisposing factors for NSCLC [3, 4]. About 95% of patients with LC are current or former smokers. Lung cancer is an invasive and heterogeneous disease. This cancer is one of the fastest growing cancers; the average time for its development is three to five months [5]. As reported in 2014, 23% of all cancers are LC [6]. Two essential characteristics of highly malignant LC are:
The tendency of cells to metastasize. In the process of metastasis, a number of signals are activated that produce continuously free radicals, including superoxide (O2•–), radical hydroxyl (OH), and hydrogen peroxide (H2O2), having several stages: epithelial–mesenchymal transition (EMT), migration to the tissue and angiogenesis [7]. The growth and survival of a cell depend on reactive oxygen species (ROS) homoeostasis. Reactive oxygen species homoeostasis is vital for healthy cells. A moderate increase in ROS can enhance the proliferation and differentiation of cells, while overproduction of ROS damages the cells. Therefore, the production of oxidants or the reduction of superoxide dismutase (SOD) and glutathione peroxidase (GPx) may cause the activation of oncogenes, abnormal metabolism, mitochondrial dysfunction, or the production of cytokines and inflammatory factors. Stimulatory signals causing chronic irritation, infections or inflammation are some of the most important causes of tumor progression [7, 8].
Resistance to treatment. In spite of huge advances in LC treatment over the past three decades, the current 5-year survival for LC is only 18% [9]. Even though multiple approaches are recommended for LC treatment such as surgery, chemotherapy, targeted therapy, immunotherapy, and radiotherapy, the mechanisms involved in the resistance of some types of LC cells to treatment are still not fully understood [10, 11].
Some lipid lowering agents such as statins and PCSK9 inhibitors are shown to have preventive effects on cancer development and incidence [12–16]. Statins are still the first-line medications for cholesterol lowering [17] because they are inhibitors 3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMG-CoA), which is a key enzyme in rate limiting synthesis of cholesterol, particularly in the liver cells [18]. However, these drugs have several pleiotropic and lipid-independent actions [19–24]. Statins used as lipid lowering agents include lovastatin, simvastatin (inactive forms), pravastatin, atorvastatin, fluvastatin (active forms) rosuvastatin and pitavastatin [17].
Recent studies have demonstrated that statins can be useful in preventing or treating cancers, due to interference with vital cell functions such as proliferation and differentiation. They can induce cell death via two mechanisms: apoptosis and necrosis in different cell lines [25]. The induction of apoptosis in cancer cells is one of the functional mechanisms by which chemotherapy drugs work and is essential for treating cancer [26]. The growth inhibitory effects and the pro-apoptotic properties of statins have attracted special attention for their treatment of various cancers, especially LC. This was the reason to review the effects of different statins in LC treatment.
Statins
Statins are the most effective low-density lipoprotein (LDL) cholesterol-lowering drugs. They are structural analogs of HMG-CoA, which competitively inhibit the enzyme HMG-CoA reductase [27]. HMG-CoA reductase is a key enzyme and regulator of the cholesterol synthesis pathway that catalyzes the conversion of HMG-CoA to mevalonate and ultimately to cholesterol. The result of inhibiting this enzyme is the inhibition of liver cholesterol production. The compensatory response to decreased hepatic cholesterol is an increase in the expression of LDL receptors on the surface of hepatocytes, resulting in increased clearance of cholesterol rich LDL particles from the blood and a decrease in their plasma levels. The major application of statins is to prevent the atherosclerotic disease [28].
Statins have lipid-lowering properties but also different side effects. Hydrophilic statins, such as pravastatin and fluvastatin, have less influence on cell membrane permeability, resulting in less pleiotropic properties and fewer adverse effects on the muscular system and central nervous system. Lipophilic statins, such as simvastatin, lovastatin, and atorvastatin, have more pleiotropic properties and side effects [29].
Lovastatin is an inactive lactone prodrug causing proteasome inhibition. It is a fungal metabolite isolated from cultures of Aspergillus terreus. Simvastatin is the chemically modified form of lovastatin [30]. Fluvastatin is the first fully synthetic HMG-CoA reductase inhibitor. The characteristics of fluvastatin include minimal systemic exposure, short half-life, and high affinity to protein binding [31]. There were some reports that fluvastatin causes less rhabdomyolyses when compared with other statins [32]. It is absorbed from the gastrointestinal tract and metabolized into the 6-hydroxy and N-desisopropyl fluvastatin by cytochrome P450 (CYP) 2C9 in the liver [33]. In some countries fluvastatin was prescribed more frequently than other statins as a first choice for treatment of hypercholesterolemia [34]. Rosuvastatin is one of the new generation of fully synthetic HMG-CoA reductase inhibitors with some unique properties. It has low lipophilicity and high protein binding affinity [35]. Rosuvastatin has a relatively long elimination half-time and has low systemic bioavailability [36]. Since it contains a polar methyl sulfonamide group, rosuvastatin interacts with HMG-CoA reductase in hepatocytes. Similarly to other statins, rosuvastatin is extensively prescribed for reduction of atherogenic plasma lipoproteins, primarily LDL and LDL cholesterol [37]. Pravastatin – the most hydrophilic drug – is relatively soluble in water and methanol [38]. Although it is quickly absorbed following oral administration, it has low bioavailability due to its ineffective absorption [39]. Pravastatin clearance is performed by the liver and kidneys and it has low affinity to protein binding [40]. Pravastatin is selectively taken up by the sodium-independent organic anion transporter protein-1B1 (OATP1B1), which is expressed in hepatocytes [41]. Pitavastatin is another member of the statin family with the following structure: heptanoate as the basic structure; a core quinoline ring, and side chains that include fluorophenyl and cyclopropyl moieties [42].
Mechanism of antitumor action of statins
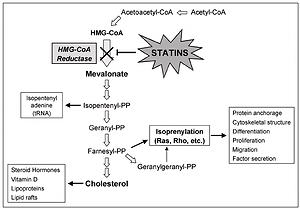
Several studies have indicated that statins can cause induction of apoptosis in different tumor cells. Possible mechanisms of induction of apoptosis by statins are due to their role in inhibiting HMG-CoA reductase and mevalonate synthesis. Mevalonate, in addition to cholesterol, is a precursor of isoprenoid molecules such as farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP). Binding of these molecules to some cellular proteins including G membrane proteins and isoprenylation of these proteins are essential for their function [43, 44]. Ras and Rho are among the most well-known isoprenylation proteins involved in the transfer of important cellular signals for transcription of genes associated with cell survival activities, including proliferation, differentiation and apoptosis. Statins have important effects on the cell because of changing the function of these proteins (Figure 1).
In various studies, increased expression of the pro-apoptotic molecules Bak, Bim, and Bax, decreased expression of apoptosis inhibitor molecules such as Bcl-2 and activation of caspases have been observed after statin therapy. In mammalian cells at least one of the main proteins – BH123, BAX and BAK – is needed in apoptosis using the intrinsic pathway. Activation of BAX and BAK proteins is dependent on the activation of BH3 pro-apoptotic proteins [45]. BAK and Bax activated by cytosol enter the mitochondrial outer membrane and disrupt the membrane permeability.
Bcl-2 family apoptosis inhibitor molecules prevent their oligomerization and activation by binding to the BH3 region in pro-apoptotic molecules [46]. Increasing Bcl-XL or Bcl-2 expression effectively inhibits apoptotic responses to cytotoxic stimuli by inhibiting ROS production, by blocking mitochondrial permeability pores, and by preventing cytochrome C release. In addition to its anti-apoptotic effect on mitochondria, Bcl-2 can inhibit pathways independent of Apaf-1 and caspase 9 and caspase-7-dependent pathways by indirect control of apoptosome activation.
By reducing the expression of pro-angiogenic factors such as vascular endothelial growth factor, statins inhibit angiogenesis in tumor tissues [47]. Vascular endothelial growth factor, an endothelial cell-specific factor, is strongly related to tumor angiogenesis and the metastatic potential of NSCLCs. Moreover, statins inhibit angiogenesis by reducing the proliferation of endothelial cells and preventing their binding to the extracellular matrix [48].
Moreover, one of the alternative effects of statins in preventing cancer is believed to be due to a mild increase in high-density lipoprotein cholesterol (HDL-C) levels, which has not been confirmed in mechanistic studies [49]. The role of serum HDL-C in the incidence of cancer or mortality remains controversial due to its various impacts on tumorigenesis [50–52].
Antitumor effects of statins in preclinical and clinical studies
Preclinical and clinical evidence has indicated that statins have anti-proliferative, pro-apoptotic and anti-invasive properties. In 2016 it was shown that lovastatin caused a decrease in viability and induction of DNA fragmentation in lung carcinoma [53]. The authors reported that high intracellular levels of the lactone caused apoptotic cell death by upregulation of COX-2 mRNA and protein, as well as increased formation of peroxisome proliferator activated receptor γ (PPARγ)-activating PGD2. Lovastatin can also stimulate tumor apoptosis in squamous cell carcinomas (SCC) by two pathways: 1) stimulation of the integrated stress response (ISR) and 2) by inhibition of ligand induced activation of the epidermal growth factor receptor (EGFR) [54], which resulted in stabilized end-stage disease in 23% of SCC patients treated with lovastatin [55].
The effect of combining lovastatin with gefitinib, a tyrosine kinase inhibitor (TKI), on gefitinib-resistant LC cell lines was also studied. The results showed that gefitinib plus lovastatin considerably improved cell growth inhibition and cytotoxicity in gefitinib-resistant NSCLC cells when compared with gefitinib alone. This could suggest that the antitumor effect is related to the up-regulation of cleaved caspase-3, poly (ADP-ribose) polymerase (PARP) and Bax, and down regulation of Bcl-2 [56]. The potential of lovastatin to induce liver kinase B1(LKB1) and AMPK as a highly conserved heterotrimeric kinase complex activity was investigated as well [57]. The effects on various metabolic stress pathways including the LKB1/AMPK pathway improve the capacity of lovastatin to act synergically with gefitinib in SCC cells.
The inhibitory effect of lovastatin on Ras farnesylation was investigated as well [58]. In human lung adenocarcinomas Kras alleles are activated by mutations and the inhibition of this has an important role in LC prevention. Some enzymes such as farnesyltransferase and inhibitors of HMG-CoA reductase inhibit Ras farnesylation [59]. It has been shown that lovastatin can destroy the formation of the tobacco-specific nitrosamine-induced lung tumors in a dose-related way without the presence of mutated ras or changes in Kras expression. The protective effect of lovastatin on normal cells against cisplatin (CDDP) cytotoxicity was assessed too [60]. CDDP is a usual chemotherapeutic drug used for the treatment of numerous solid tumors. The authors reported that lovastatin at the pharmacologically relevant concentration per se induced DNA damage, autophagy and oxidative stress in a human lung adenocarcinoma cell line but not in normal human mesothelial primary cells. Therefore, in cancer patients, lovastatin might improve the therapeutic index of cisplatin-based chemotherapy. Zhang et al. investigated whether lovastatin inhibits proliferation due to minichromosome maintenance (MCM) 2 in human NSCLC [61]. They measured the effects of lovastatin on cell anti-proliferation, cell cycle progression and apoptosis in NSCLC cells. Recent studies have also shown that a novel anti-proliferation mechanism of MCM2 could be used for potential therapeutic effects in NSCLCs by knockdown of MCM2 inhibiting not only the G1/S cell cycle but also by activation of apoptosis. Lovastatin has been used in vivo as an inhibitor of tumor progression in various tumor models such as colon, pancreas, and melanoma [62–64]. It has been shown that high doses of lovastatin (0, 25 and 50 mg/kg 3 times a week) were able to inhibit lung metastases in vitro in syngeneic BALB/c mice [65]. In this study, the effects of lovastatin in mammary cancer cells stopped the G1 phase and decreased S and G2/M phases of the cell cycle.
Several investigations in vitro and epidemiologic studies have indicated antitumor effects and protective effects of atorvastatin decreasing the risk of LC [66, 67]. Platinum-based chemotherapy is often used for treatment of progressive NSCLCs. Previous studies have shown that atorvastatin when combined with carboplatin had better effects on the apoptosis of NSCLC when compared with either drug applied as monotherapy [56]. It has been shown that treatment of LC using atorvastatin in combination with carboplatin decreased the progression of xenograft A549 tumors in mice by suppressing AKT activity and improved the survival when compared with carboplatin treatment as monotherapy [68, 69].
Lu et al. investigated the synergistic action of Polyphenon E (PPE, green tea polyphenol) and atorvastatin on the inhibitory effect in a mouse model and in vitro [70]. Their results indicated that PPE or atorvastatin alone was not effective in suppressing lung carcinogenesis. The combination of PPE and atorvastatin in low amounts reduced both the tumor growth and tumor expansion.
This synergistic effect of PPE plus atorvastatin was associated with improved apoptosis as determined by the terminal deoxyribonucleotide transferase based nick-end labeling assay. The combination of PPE plus atorvastatin had an additional effect in decreasing the antiapoptotic protein Mcl-1 level and the effect on caspase-3 and cleaved poly (ADP)-ribose polymerase level, which was different from monotherapy with each of these substances. This study showed that PPE plus atorvastatin synergistically inhibited 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced lung tumorigenesis in mice and the growth of LC H1299 and H460 cells, probably by increased apoptosis [70].
Recent studies have shown that cancer cells include poor antioxidant defense. Simvastatin, due to oxidative stress metabolites and SOD2 expression, causes an increase in the regulation of A549 protein and inhibition of cancer cell development [71]. The beneficial effects of treatment with a combination of cisplatin and simvastatin on patients with SCC have also been demonstrated. Such treatment reduced renal toxicity in these patients, and even increased their life span [72]. It is well known that smoking causes DNA damage and LC. The level of metalloproteins in the plasma is increased, and the level of RAS proteins is decreased, which results in the activator protein-1 (AP-1), by PI3K/Akt, JNK, p38 MAPK, and ERK signaling pathways [73]. SIM-induced G1 cell cycle stopping in NCI-H460 cells is mediated by the upregulation of p16 and p27 proteins, and induces blockade of cyclin D-Cdk4 expression. All these phenomena result in the inhibition of proliferation of NSCLC and reduction of the number of cells in S phase and G1 of the cell cycle. This suggests that the cell cycle-suppressing activity of simvastatin is associated with its anti-cancer properties [74]. BIM is an active agent of apoptotic protein, and it has been reported that simvastatin causes expression of BIM, inhibition of ERK protein signals, and the death of apoptotic cells in H1975 cell culture. These effects were mediated by expression of the BIM protein. Epithelial to mesenchymal transition is pivotal for driving metastasis and recurrence in LC. Since some in vitro studies have shown that statins suppress this transition by inactivating mutant p53 functions, it has been suggested that simvastatin suppresses it and changes the prognosis of patients with lung adenocarcinoma in a p53 mutation-dependent manner [75]. Finally, it seems that simvastatin might even cause cancer cells to disappear [76].
Preclinical data have demonstrated the anti-tumor effects of fluvastatin in various cancers. It has been shown that fluvastatin synergistically enhances the anti-proliferative effect of gemcitabine in human pancreatic cancer cells [77]. Furthermore, fluvastatin in combination with cisplatin showed synergistic anti-tumor activity in epithelial ovarian cancer cells by inhibiting the Ras pathway [78]. Previous studies have already indicated that fluvastatin can have prophylactic effects in cancer. In a study on renal cancer, fluvastatin inhibited renal cancer cells time- and dose-dependently by induction of cell cycle arrest and apoptosis in the cells. It was shown that fluvastatin influences this process by up-regulation of p21 and p53 in this type of cancer. Moreover, it can prevent angiogenesis, invasion, and metastasis by decreasing phosphorylation of rac1 in renal cancer cells [31]. Recently, a significant body of evidence has demonstrated the anti-proliferative and chemotherapeutic effects of fluvastatin in LC. After it had been shown that HMG-CoA reductase is overexpressed in human lung adenocarcinoma when compared with normal tissues, fluvastatin, an inhibitor of HMG-CoA reductase, suppressed NSCLC cell growth and induced apoptosis by inhibiting the HMG-CoA reductase-driven Braf/MEK/ERK1/2 and Akt signaling pathways. It also attenuated tumor growth in 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis [79].
Xu et al. reported that fluvastatin significantly reduced in vitro cell proliferation and metastasis of aggressive human NSCLC cells and this effect was time- and dose-dependent. Furthermore, they found that fluvastatin significantly suppressed the in vitro tumor progression probably by down-regulation of SATB1 (Special AT-rich sequence binding protein 1) expression in the human NSCLC cell line H292 [80]. Yang et al. observed that fluvastatin significantly reduced bone metastasis of lung adenocarcinoma in a nude mouse model. They suggested that fluvastatin mainly inhibited bone metastasis by induction of a p53-dependent autophagy process in lung adenocarcinoma cells [81].
Levine and Cagan observed in a Drosophila LC model that oral administration of MEK inhibitor (trametinib) and fluvastatin blocked Ras and PI3K signaling pathways, which decreased survival in the models. The combination of these drugs showed synergistic suppression of tumor progression and viability in the models. This combination also exhibited a cytotoxic effect on human lung adenocarcinoma cells [82]. All these data suggest that fluvastatin could be considered as a novel chemotherapeutic agent in cancer, particularly in LC.
Previous studies suggested that rosuvastatin might have an important role in therapy of various cancers. In a randomized clinical trial in women with breast cancer, it was found that rosuvastatin prevented the development of chemotherapy-induced cardiotoxicity [83]. In addition, rosuvastatin enhanced the anti-cancer effect of dasatinib in hepatocellular carcinoma via inhibiting the FAK/Src signaling pathway [84]. Yokohama et al. found that rosuvastatin prevented hepatocellular carcinogenesis and improved the hepatic background by down-regulation of the mRNA level of pro-inflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin 6 (IL-6) and transforming growth factor β1 (TGF-β1) in mice [85]. In melanoma cells, rosuvastatin dose-dependently reduced cell proliferation. Rosuvastatin also down-regulated ki67 expression in melanoma cells in prolonged incubation [86]. Some studies demonstrated that rosuvastatin can reduce the effects of oxidative stress and DNA damage by induction of antioxidant defense enzymes such as γ-glutamylcysteine synthetase in the cells [87].
Janakiram et al. reported that rosuvastatin alone or in combination with difluoromethylornithine significantly decreased polyamine content in the cells and increased intra-tumoral NK cells expressing perforin and IFN-γ when compared to untreated colon tumors [88]. Recently, a large body of evidence has suggested the anti-tumor effect of rosuvastatin in LC. Liu et al. found that statin treatment significantly decreased LC risk in chronic obstructive pulmonary disease (COPD) patients and the effect was dose-dependent. Nevertheless, lovastatin and fluvastatin did not show any significant reduction in LC risk while not only rosuvastatin, but also simvastatin, atorvastatin, and pravastatin significantly decreased LC risk [89]. Simvastatin and rosuvastatin significantly inhibited cell proliferation by suppression of RAS protein, matrix metalloproteinases (MMP-2/9) and NF-κB-p65 expression in LC tissues, and this effect was dose-dependent [90]. However, there are some conflicting data as well. Lai et al. found that rosuvastatin with cumulative use duration > 12 months was associated with 2.8-fold increased risk of LC in Taiwan women when compared with those who were not treated with statins [91]. Some studies indicated that liposomal pravastatin treatment effectively inhibited the production of several pro-inflammatory/pro-angiogenic mediators involved in inflammation and angiogenesis in cancer [92]. Inconsistently with previous results, a small randomized phase II clinical trial on thirty patients concluded that addition of pravastatin to epirubicin, cisplatin, and capecitabine did not improve the clinical outcome of the enrolled patients with advanced gastric cancer [93]. A more recent study also showed that pravastatin did not significantly improve clinical outcome in patients with advanced gastric cancer (stage III or IV) [94].
A meta-analysis including 13 724 patients demonstrated that pravastatin treatment was not associated with cancer risk. Pravastatin did not influence overall survival of the patients either [95]. Asakage et al. found that pravastatin decreased cell proliferation and growth by cell cycle arrest in G1 phase due to depleting cyclin D, cyclin E and cyclin-dependent kinase 2 expression levels in endothelial cells [96]. With regards to LC, a multicenter phase III randomized double-blind placebo controlled clinical trial, which investigated the effects of pravastatin added to standard chemotherapy in SCC, showed that pravastatin did not bring any benefit in the patients. The 2-year overall survival (OS) and progression-free survival (PFS) rates of pravastatin and placebo did not show any significant difference [97]. In a recent review on pravastatin in LC, it was concluded that pravastatin did not have any beneficial effects in SCC [98]. Taras et al. demonstrated that pravastatin significantly reduced tumor progression and lung invasion in rat hepatocellular carcinoma. They showed that pravastatin inhibited tumor metastasis to the lung by reducing liver matrix metalloproteinase-9 (MMP-9) activity and suppression of MMP2, which was mainly attributed to down-regulation of MMP-14 in hepatocellular carcinoma [99]. In the animal study of Ferreira et al., atorvastatin and pravastatin slightly reduced inflammation and oxidative stress in mice exposed to cigarette smoke in comparison to controls. Rosuvastatin showed the highest anti-inflammatory effect and simvastatin achieved the best anti-oxidative response [100]. As mentioned above in the section on rosuvastatin, Liu et al. found that statin significantly reduced LC risk in chronic obstructive pulmonary disease patients and this effect was dose-dependent. They reported that pravastatin was significantly associated with a decreased LC risk [89]. Pravastatin showed a marginal benefit in cancer therapy and further investigation will be needed to explain the exact role and possible effect of pravastatin in cancer.
Otahal et al. found that pitavastatin and fluvastatin induced cell death in EGFR TKI resistant NSCLC cells lines A549, Calu6 and H1993 in vitro. Co-treatment of cells with pitavastatin and the EGFR TKI erlotinib resulted in synergistically enhanced cytotoxicity when compared to pitavastatin monotherapy overcoming erlotinib resistance in K-ras mutated NSCLC and relying only on apoptosis [101]. Hu et al. demonstrated that pitavastatin can inhibit LC cells and angiogenesis, which was achieved by the induction of apoptosis and inhibition of proliferation of LC cells and human lung tumor-associated endothelial cells. They also demonstrated that pitavastatin acted on LC cells and human lung tumor-associated endothelial cell by suppressing prenylation-dependent Ras/Raf/MEK and PI3K/Akt/mTOR signaling [102].
In a meta-analysis on the antitumor effects of statins, it was found that patients who suffered from lung tumors and used statins had better overall survival than other patients who did not use statins. Subgroup analyses were performed as well, indicating that use of statins in stage IV LC was associated with a better survival rate when compared to other stages [103].
In a case-control study Khurana et al. evaluated the anti-tumor effects of statins in patients with LC. Based on the results of this study, the use of statins reduced the risk of LC in the veteran population [104].
In a study by Omori et al. on patients with NSCLC, the results demonstrated that the use of statins with nivolumab improved the treatment of patients when compared to those who were not treated with statins [105].
Conclusions
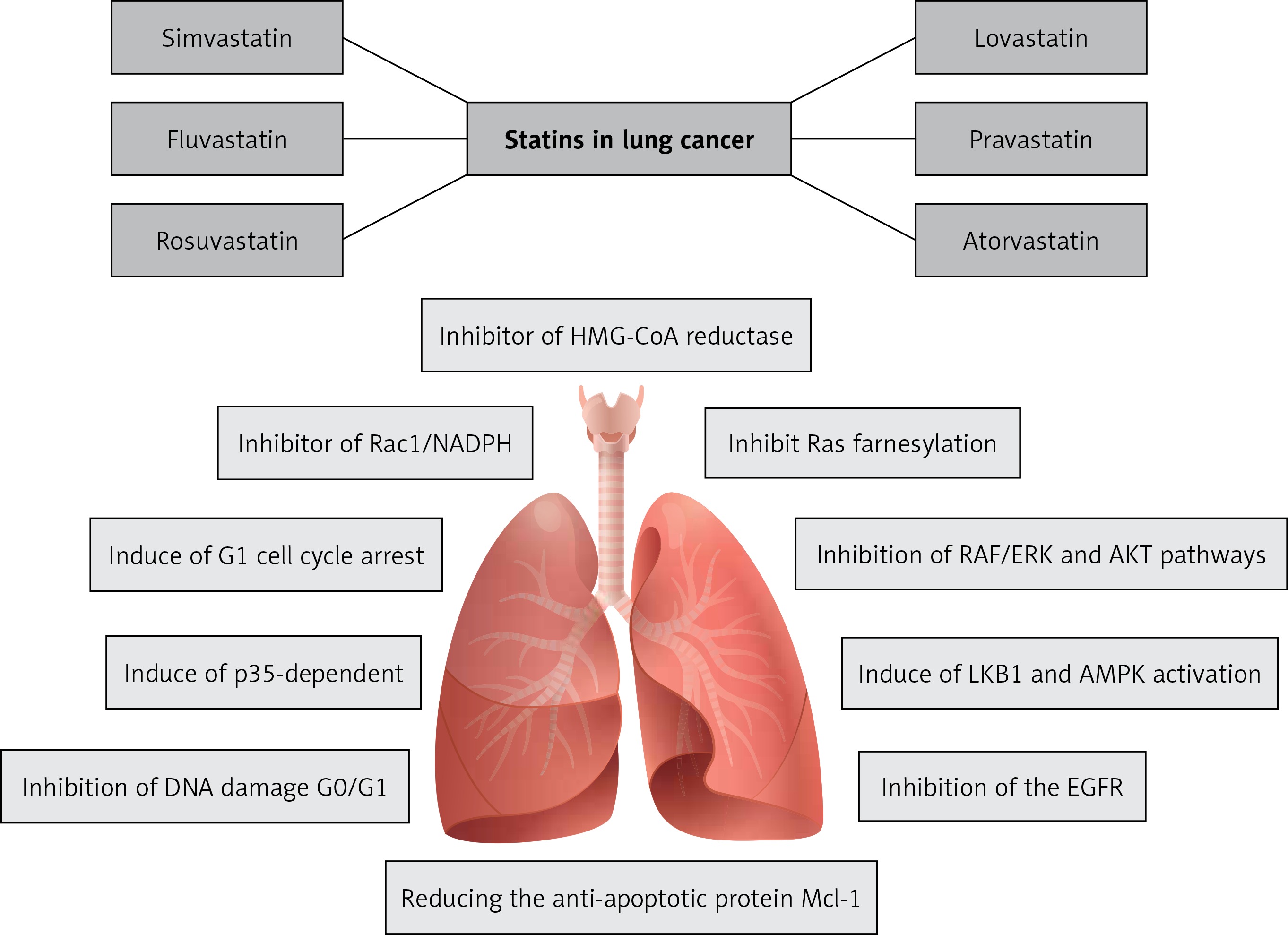
This review presents the possible mechanisms of statins including lovastatin, atorvastatin, simvastatin, fluvastatin, rosuvastatin, pitavastatin and pravastatin for the prevention and treatment of LC (Figure 2). Pre-clinical studies and animal models of cancer therapy indicate the antitumor effects of statins on various types of cancer, especially LC. On the basis of these studies, some potential antitumor properties of statins have been identified and reported. However, the molecular pathways and intracellular signaling cascades involved in the inhibition of cancer are very broad and complex, and there is a clear need for further studies.
Figure 2
The most important effects of statins on lung cancer: inhibiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, stopping the mevalonate production from HMG-CoA and reducing mevalonate level, inhibiting myocardial Rac1-GTPase activity in order to suppress the NADPH oxidase activity and ROS generation. Also statins are inhibitors of farnesyltransferase (FT) proteins, RAF/ERK and AKT pathways, inhibitors of the EGFR and anti-apoptotic protein Mcl-1 in animals and humans. Additionally, they have effects on cellular energy homeostasis by inducing LKB1 and AMPK activation, and by inducing p53 pathway and G1 cell cycle arrest, which prevents DNA damage
Epidemiological studies have also illustrated that there might be a correlation between the use of statins and the reduction of LC risk. However, because clinical studies are very limited, the usefulness of statins to help treating LC cannot be definitively assessed. Phase II and III clinical studies on a large number of patients are needed to demonstrate the possible antitumor effects of statins in different types of lung tumors. It is quite obvious that statins will never be drugs for LC treatment. Nevertheless, if proven so, they might be a useful supplemental tool and an additional treatment to all the other well-established therapies for LC management (Table I) [106–113].
Table I
Statins for treatment of lung cancer
| Type of statin/doses of agent | Mechanism | Investigated cell type | Result | Ref. no. |
|---|---|---|---|---|
| Lovastatin: | ||||
| 50 μM | COX-2 activation of PPARγ | A549 and H358 lung carcinoma cells | Human lung cancer cell death | [53] |
| 0, 160, or 400 ppm | Inhibited ras farnesylation | NCI-H125, H292, H441, H460, and H661 | Lovastatin inhibited the growth in vitro of mouse | [58] |
| 5 μM in A549, 1 μM in H460 | Inhibition of both RAF/ERK and AKT pathways | Human NSCLC lines | Overcame gefitinib resistance | [56] |
| 5 μM | Induction of the ISR and inhibition of the EGFR | SCC | Apoptosis in SCC cells | [54] |
| 1–25 μM | Induced LKB1 and AMPK activation | A549, HeLa, SCC9, SCC25 cell lines | Regulating lovastatin-induced cytotoxicity | [57] |
| 2.5–20 μM | Anti-proliferation mechanism of MCM2 | Human NSCLC | Activation of apoptosis | [61] |
| 20 μM | A p53-independent mitochondrial-mediated apoptotic mechanism | BJMC3879 mammary adenocarcinoma cell line | Tumor volumes were significantly reduced | [65] |
| 2 μM | Inhibition of DNA damage-dependent G0/G1 | MCF-7 and A549 | Improvement of the therapeutic index of cisplatin-based chemotherapy | [60] |
| Atorvastatin: | ||||
| 1–10 μM in vitro | Reduced the growth of xenograft and inhibition of AKT activity | A549 and H1299 | Atorvastatin overcame carboplatin resistance in lung cancer | [69] |
| 5 μM in vitro 10 mg/kg in vivo | Suppression of Rac1/NADPH oxidase activity | Non-small cell lung cancers (NSCLCs) A549 cells | Inhibit VEGF expression via inhibition of ROS production | [106] |
| 1 or 5 μM in vitro 1 or 10 mg/kg in vivo | Suppression of AKT and ERK activity | A549 and Calu1 cells | Atorvastatin + gefitinib inhibited proliferation and promoted cell apoptosis | [107] |
| 1–8 μM in vitro 200 or 400 ppm in vivo | Reducing the anti-apoptotic protein Mcl-1 and increasing cleaved caspase-3 | Human lung cancer H1299 and H460 cell lines | PPE plus atorvastatin synergistically inhibited lung tumorigenesis | [70] |
| Simvastatin: | ||||
| 50 μM in vitro | Akt signaling | A549 lung carcinoma cells | Inhibited the proliferation of A549 lung cancer cells | [53] |
| 2.5–30 μM | Protein RAS, matrix metalloproteinases and NFκ-B | Lung neoplastic cells | Inhibition of cancer growth in vitro | [58] |
| 20 μM | Activation of Bax and down-regulation of BCL-2 gene expression | MCF7 human breast cancer cells | Inhibits cancer cell growth by inducing apoptosis | [108] |
| 1, 10, 20, 100 μM | Induced G1 cell cycle arrest | NCI-H460 cells | Anti-cancer properties | [57] |
| 2 μM | Expression of BIM and inhibition of ERK | H1975 cells | Activation of apoptosis | [76] |
| Fluvastatin: | ||||
| 0, 5, 10, 20, 40 μM | Down-regulating of STAB1 expression level via the Wnt/β-catenin pathway | Human NSCLC cell line H292 | Reduced cell proliferation and invasion | [80] |
| 10 μM | Inhibited bone metastasis through induction of p53-dependent autophagy process | A549 and SPC-A-1, and animal model | Reduced bone metastasis | [81] |
| 1–3 μM | In combination with trametinib, blocked Ras and PI3K signaling pathways | Drosophila models and A549 cell line | Showed synergistic suppression of tumor progression and viability | [82] |
| Pravastatin: | ||||
| NA | Chemopreventive effect against lung cancer | Lung cancer in COPD patients | Decreased lung cancer risk remarkably | [89] |
| 40 mg | NA | Multicenter, phase III, randomized, double-blind, clinical trial | Did not present any benefit in the patients | [97] |
| Rosuvastatin: | ||||
| NA | Chemopreventive effect against lung cancer | Lung cancer in COPD patients | Decreased lung cancer risk remarkably | [89] |
| 1.25, 2.5, 5, 10, 30 μM | Inhibited cell proliferation | Lung cancer tissues | Suppression of RAS protein, matrix metalloproteinases (MMP-2/9) and NF-kB-p65 expression | [90] |
| Pitavastatin: | ||||
| 10 μM | Induced cell death in EGFR TKI resistant cells | A549, Calu6 and H1993 in vitro | Induced cell death in EGFR TKI resistant NSCLC cells lines | [109] |
| 1–5 μM | By suppressing prenylation-dependent Ras/Raf/MEK and PI3K/Akt/mTOR signaling | Human lung tumor-associated endothelial cell | Induction of apoptosis and inhibition of proliferation | [110] |
[i] COX – cyclooxygenase, NSCLC – non-small cell lung cancer, SCC – squamous cell carcinomas, COPD – chronic obstructive pulmonary disease, PPARγ – peroxisome proliferator activated receptor γ, EGFR – epidermal growth factor receptor, ISR – integrated stress response, PPE – green tea polyphenol, NM – not mentioned.