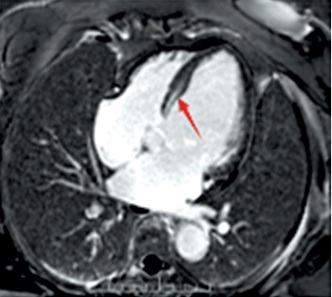
A case study of a classic cardiac phenotype resulting from a rare missense mutation in the LMNA gene is presented to underscore the vital significance of comprehensive genetic assessment in the context of specific clinical manifestations. The patient, a 67-year-old woman, was hospitalized in May 2025 due to a one-month history of “paroxysmal palpitations with associated dizziness and weakness”. Of particular note was her family history: her mother succumbed to coronary artery disease, and notably, her 10-year-old granddaughter had experienced recurrent unexplained syncopal episodes, strongly indicating a potential hereditary predisposition across generations. On admission examination, the cardiac border was found enlarged to the left and downward. Occasional premature contractions were heard on auscultation. Holter monitoring revealed frequent non-sustained ventricular tachycardia (NSVT) (83 episodes/24 h), marked heart rate variability (maximum 210 bpm, minimum 38 bpm), and sinus pauses (longest 2.8 s), consistent with the typical arrhythmic phenotype of LMNA-related cardiomyopathy. The coexistence of atrioventricular conduction abnormalities and ventricular arrhythmias represents an important marker of a sudden cardiac death risk in patients with LMNA variants, and constitutes a clear indication for implantable cardioverter-defibrillator (ICD) implantation. Cardiac ultrasound demonstrated marked left ventricular enlargement, a blunted apical cone, diffuse and dyssynchronous wall motion, and significantly impaired left ventricular systolic function (LVEF approximately 35%), the right ventricle was normal in size (RV 18 mm, RVOT 28 mm) and function. To precisely quantify the cardiac structure and function and investigate the etiology, the patient had cardiac magnetic resonance (CMR) imaging performed. As shown in Figure 1, CMR imaging demonstrated a marked left ventricular dilatation with a severely reduced global systolic function. The key finding was patchy mid-myocardial late gadolinium enhancement (LGE) involving the left ventricular apex and lateral wall, comprising approximately 15% of left ventricular myocardial mass, with a non-ischemic distribution pattern consistent with myocardial fibrosis. To comprehensively rule out coronary artery disease – the most common cause of left ventricular dysfunction – and confirm the diagnosis of non-ischemic cardiomyopathy, the patient underwent coronary angiography, which revealed non-obstructive coronary artery disease (CAD) with all stenoses < 50%. This finding definitively ruled out clinically significant obstructive coronary artery disease. Given the patient’s presentation of “dilated cardiomyopathy with high-burden ventricular tachycardia and conduction abnormalities (frequent premature ventricular contractions)” and a family history of syncope in younger relatives, hereditary cardiomyopathy – particularly laminoproliferative disease – was highly suspected clinically. Following informed consent, peripheral blood was collected and subjected to next-generation sequencing using a hereditary cardiomyopathy/arrhythmia-related gene panel (Version V3). Genetic analysis revealed a heterozygous missense mutation in exon 3 of the LMNA gene: c.565C>T (p.Arg189Trp), as detailed in Table I. This variant causes substitution of arginine at position 189 with tryptophan in the encoded protein. According to the American College of Medical Genetics and Genomics guidelines [1], this variant is classified as likely pathogenic based on the following rationale: the site exhibits high conservation across multiple species; arginine is a crucial polar amino acid, and its replacement with the hydrophobic tryptophan is predicted to adversely affect protein structure/function; the mutation exhibits extremely low frequency in normal population databases; it demonstrates high concordance with the patient’s phenotype (clear features of LMNA-related cardiac pathology).
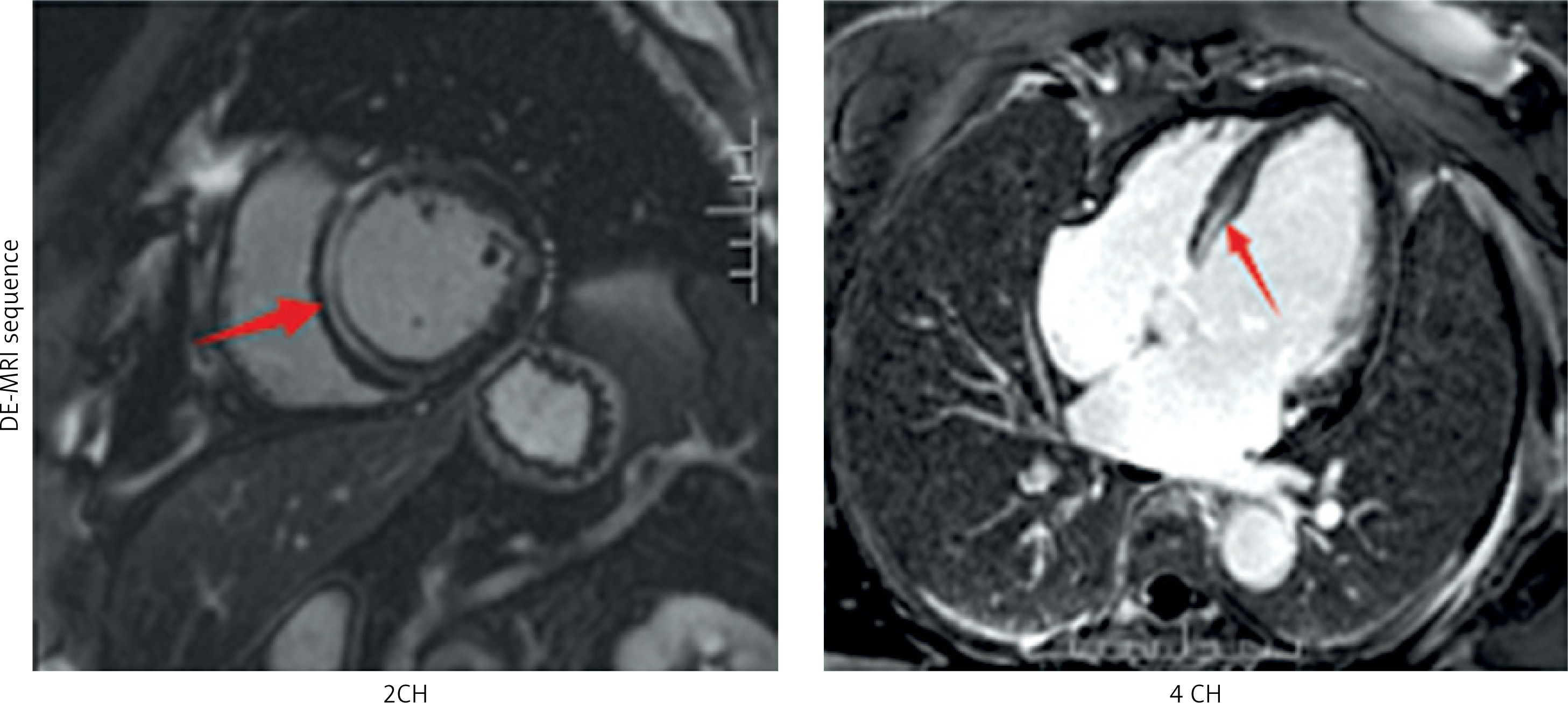
Table I
Genetic Testing Analysis Report
[i] Note: Prediction: REVEL, a protein function prediction software: D: Damaging; LD: Likely Damaging; U: Uncertain; LB: Likely Benign; B: Benign; Score: Bayesian framework scores/Bayesian scores for uncertain sites and their corresponding probability of pathogenicity: < 0 (0.1%); 0 (10%); 1 (18.8%); 2 (32.5%); 3 (50%); 4 (67.5%); 5 (81.2%); ≥ 6 (≥ 90%).
The LMNA gene encodes lamin A/C, and its mutations can cause a group of diseases known as laminopathies [2, 3]. The cardiac phenotype is characterized by dilated cardiomyopathy with early-onset conduction system disorders (such as atrioventricular block) and malignant ventricular arrhythmias, often exhibiting age-dependent expression [4, 5]. This patient developed severe symptoms at the age of 67, while her granddaughter experienced syncope at the age of 10, perfectly illustrating this feature. Non-ischemic delayed enhancement on CMR is a common pattern of fibrosis in LMNA cardiomyopathy [6], associated with arrhythmogenic matrix [7]. The identification of this gene mutation carries significant clinical implications. It immediately suggests the diagnosis in this patient from “idiopathic” to “genetically confirmed”. More importantly, it initiated a cascade of familial screening. Following our recommendation, the patient’s children and symptomatic granddaughter have undergone genetic testing at this locus alongside cardiac evaluation (including ECG, Holter monitoring, and echocardiography). Family members harboring this mutation require regular monitoring even without symptoms, with early discussion of primary prevention via implantable cardioverter-defibrillator (ICD) placement [8, 9] as LMNA mutation carriers face a high risk of sudden cardiac death [7, 10]. For patients meeting the “dilated cardiomyopathy with significant conduction abnormalities or ventricular arrhythmias” pattern [11], especially those with any suggestive family history, LMNA genetic testing should be incorporated into the early diagnostic workflow. This approach not only confirms diagnosis but also enables risk stratification and preventive management, potentially saving the lives of affected family members [12].