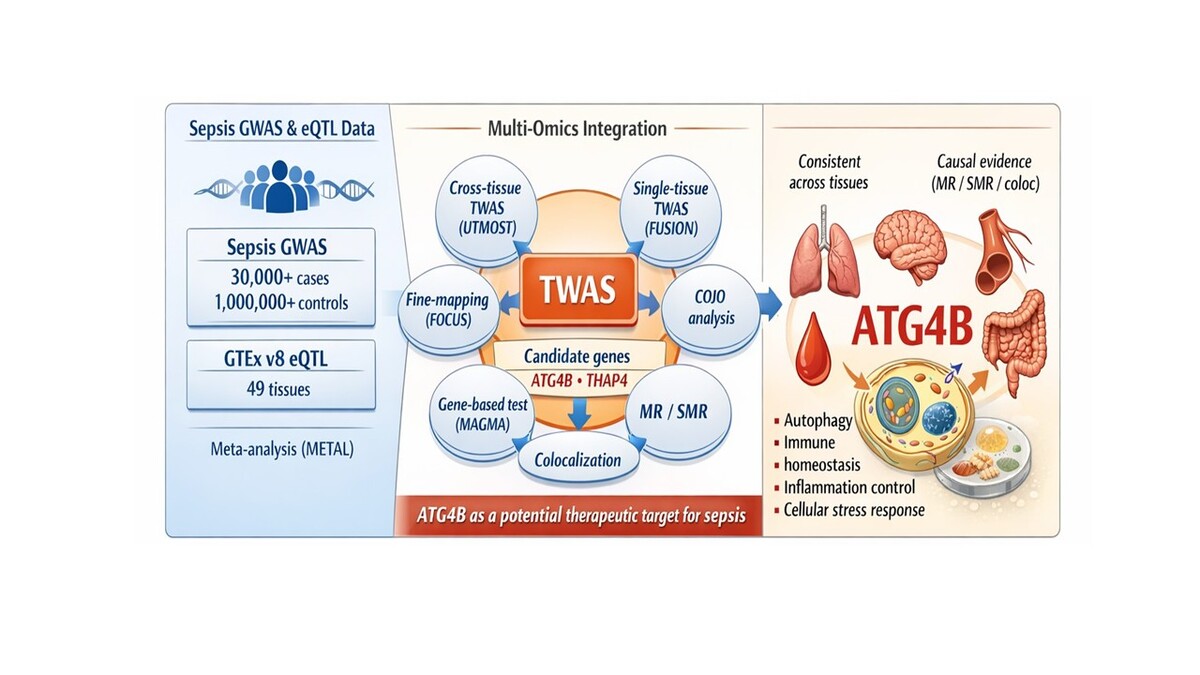
Sepsis is a life-threatening syndrome resulting from a dysregulated host response to infection, leading to widespread inflammation, tissue injury, and multi-organ dysfunction [1]. It remains a significant global health burden, contributing substantially to morbidity and mortality [2]. The pathogenesis of sepsis is highly complex, involving intricate interactions between host immune responses, genetic predispositions, and environmental factors [3]. Despite extensive genome-wide association studies (GWAS) identifying genetic variants associated with sepsis susceptibility, the genetic basis of the disease remains incompletely understood due to its polygenic nature and the limitations of traditional single-layer approaches [4]. To address this, we employed a multi-omics integration strategy combining GWAS, eQTL, and transcriptome-wide association studies (TWAS) to uncover gene-trait associations and refine candidate genes, thus enhancing our understanding of sepsis pathogenesis and informing potential therapeutic strategies.
Methods
The analysis process is illustrated in Supplementary Figure S1.
Sepsis GWAS data source
Sepsis data for our study were sourced from the GWAS Catalogue (https://www.ebi.ac.uk/gwas/home), with a specific focus on hospital admission cases. We incorporated two curated datasets: one by Butler-Laporte G, which includes 18,931 sepsis cases and 663,531 controls (GCST90270871), and another by Hamilton FW, comprising 11,643 cases and 474,841 controls (GCST90281174). To enhance statistical power and comprehensively assess the genetic associations, we merged these two GWAS datasets and performed a meta-analysis using METAL software (https://csg.sph.umich.edu/abecasis/Metal/index.html). This analysis utilised a random-effects model under the inverse variance weighting method – specifically, a multiplicative approach – to accommodate potential heterogeneity between studies.
eQTL files source
The eQTL files were derived from the GTEx V8 dataset, which provides extensive gene expression profiles across 49 distinct tissues sourced from 838 post-mortem donors (https://ftp.ebi.ac.uk/pub/data-bases/spot/eQTL/imported/GTEx_V8). Notably, sample sizes varied considerably among tissues, from as few as 73 samples in the renal cortex to up to 706 in skeletal muscle.
TWAS analyses in cross-tissue
In our cross-tissue transcriptome-wide association study (TWAS), we utilised the UTMOST framework (https://github.com/Joker-Jerome/UTMOST?tab=readme-ov-file) to assess overall gene–trait associations at the organismal level. This methodology facilitated the identification of additional genes in tissues with enriched trait heritability and improved the accuracy of gene expression imputation [5]. Furthermore, we integrated the gene–trait association signals using the generalised Berk-Jones (GBJ) test, which leverages covariance information derived from single-tissue analyses [5, 6]. Statistical significance was determined by applying a false discovery rate (FDR) correction, and associations achieving an FDR threshold of < 0.05 were considered statistically significant.
TWAS analyses in single tissue
To assess the association between gene expression and sepsis risk on a tissue-specific basis, we employed the FUSION tool (http://gusevlab.org/projects/fusion/). This approach integrates sepsis GWAS data with eQTL information from GTEx V8 covering 49 tissues, thereby allowing us to estimate the contribution of each gene to disease susceptibility. Initially, the linkage disequilibrium (LD) between the prediction model and the SNPs at each GWAS locus was calculated using European samples from the 1000 Genomes Project. Subsequently, FUSION combined several predictive models – namely, BLUP, BSLMM, LASSO, Elastic Net, and the top-1 method – to assess the overall impact of SNPs on gene expression. The model that exhibited the highest predictive performance was then chosen to determine the final gene weights. Finally, these gene weights were integrated with the sepsis GWAS Z-scores to perform the transcriptome-wide association study (TWAS). For further analysis, candidate genes were selected based on meeting two criteria: (1) a false discovery rate (FDR) of less than 0.05 in the cross-tissue TWAS analysis, and (2) an FDR of less than 0.05 in at least one tissue in the single-tissue TWAS analysis.
Fine-mapping of causal gene sets (FOCUS) analysis
In addition, we conducted a FOCUS analysis to further refine the identification of causal genes underlying the TWAS signals (https://github.com/mancusolab/ma-focus). FOCUS (fine-mapping of causal gene sets) is a probabilistic fine-mapping method specifically designed to accurately discern causal genes from TWAS data. This approach overcomes the limitations inherent in traditional methods, particularly those arising from linkage disequilibrium (LD) and the influence of pleiotropic SNPs. By modelling the correlation structure of gene expression prediction, FOCUS effectively extracts a set of candidate causal genes associated with complex traits and diseases, providing a highly credible prioritisation for downstream gene validation and functional studies. In the present study, the significance thresholds were set such that genes falling within the 90% confidence interval and exhibiting posterior probability values ≥ 0.9 were considered statistically significant [7].
Conditional and joint analysis
In FUSION, it is common to detect multiple associated features within a single locus, prompting the need to assess their conditional independence. To address this, we performed a conditional and joint (COJO) analysis using the post-processing module in FUSION [8]. This approach accounts for the linkage disequilibrium (LD) between markers, thereby providing a more nuanced understanding of the genetic architecture underlying trait variation. Post-analysis, genes that retained significance after accounting for the LD structure were deemed jointly significant, while those that lost significance were classified as marginally significant.
Gene analysis
For our gene-level analysis, we applied MAGMA software (version 1.08) using default settings. This approach aggregates SNP-level association statistics into composite gene scores, thereby quantifying the degree to which each gene is associated with the phenotype [9, 10]. For a detailed discussion of the parameter settings and methodological framework underpinning this analysis, readers are encouraged to consult the original MAGMA documentation.
Mendelian randomisation and Bayesian colocalization
We conducted Mendelian randomisation (MR) analysis using the “TwoSampleMR” R package. In this analysis, cis-eQTL SNPs were employed as instrumental variables (IVs), with gene expression serving as the exposure and sepsis GWAS as the outcome. Initially, genome-wide significant SNPs (p < 5 × 10–5) were selected, and linkage disequilibrium (LD) clumping (r2 < 0.001) was performed to retain independent variants. Due to the availability of only a single standalone IV, the MR effect was estimated using the Wald ratio, applying a significance threshold of p < 0.05. Additionally, we applied summary-data-based Mendelian randomisation (SMR) to evaluate the relationship between gene expression and sepsis risk. This method leverages the most significant cis-eQTL – identified within a ±1000 kb window around each gene and meeting the stringent significance threshold (p < 5.0 × 10–8) – to achieve greater statistical power than conventional MR when exposure and outcome data derive from large, independent samples [11]. Variants exhibiting allele frequency differences greater than 0.2 between any pair of datasets (i.e. the LD reference panel, QTL summary, and outcome summary) were excluded from the analysis. The HEIDI test was then employed to distinguish pleiotropy from linkage; SNPs with P-HEIDI values below 0.01 were removed because they were deemed likely to be reflective of pleiotropy. Both SMR and HEIDI analyses were performed using SMR software (v1.3.1). To control the false discovery rate (FDR) at α = 0.05, p-values were adjusted using the Benjamini-Hochberg method, and only associations with an FDR-corrected p-value below 0.05 and a P-HEIDI exceeding 0.01 were considered statistically significant.
Subsequently, a Bayesian colocalisation analysis was carried out using the “coloc” R package to determine whether causal variation loci overlapped between the GWAS and eQTL signals. This analysis assessed the posterior probabilities (PP) corresponding to five models. In our colocalization analysis, a posterior probability of H4 (PPH4) greater than 0.70 was used as supporting evidence for colocalisation. This threshold, which corresponds to a false discovery rate (FDR) of < 5%, strengthens the evidence for a causal relationship between the GWAS and eQTL signals.
Results
TWAS analyses in cross-tissue and single tissue
In the cross-tissue TWAS analysis, 617 genes reached nominal significance (p < 0.05) (Supplementary Table SI), among which 114 genes remained significant after adjustment for multiple testing (FDR < 0.05) (Supplementary Table SI). For the single-tissue TWAS validation, 3260 genes with FDR < 0.05 in at least one tissue were identified (Supplementary Table SII). In parallel, our FOCUS analysis pinpointed 195 genes exhibiting significant associations with sepsis (Supplementary Table SIII). These genes were prioritised based on their high posterior probability scores, which suggest their potential roles as causal drivers in sepsis pathogenesis. By applying these stringent criteria to these analyses, we ultimately identified 6 candidate genes that demonstrated robust associations across cross-tissue and single-tissue analyses. All these genes are protein-coding (LRRC66, RASL11B, ATG4B, RPL3, SGCB, THAP4) (Supplementary Table SIV).
COJO analysis
The 6 candidate genes, predominantly located on chromosomes 2, 4, and 22, were subjected to conditional and joint (COJO) analysis in their respective tissues to mitigate false positive associations that may arise from linkage disequilibrium (LD) (Supplementary Table SV). Specifically, in lung tissue, conditioning on ATG4B expression did not appreciably alter the TWAS signal for RP11–367H1.1 (Supplementary Figure S2 A). Likewise, in brain cerebellum, ING5’s association remained largely unchanged after adjustment for ATG4B (Supplementary Figure S2 B), and similar stability was observed for DTYMK in muscle skeletal (Supplementary Figure S2 C) and THAP4 in stomach (Supplementary Figure S2 D). These findings suggest that the marginally significant signals at this locus are not driven by ATG4B’s predicted expression. In brain anterior cingulate cortex BA24, conditioning on the predicted expression of both RASL11B and DANCR produced only a modest reduction in ERVMER341’s TWAS signal (Supplementary Figure S2 E), indicating that ERVMER341 retains a degree of independent association at this locus.
Analysis of MAGMA
MAGMA gene-based analysis identified 1323 significant genes associated with sepsis (FDR < 0.05) (Supplementary Table SV, Supplementary Figure S3). To further enhance the robustness of our findings, we integrated UTMOST cross-tissue results with the significant genes identified via FUSION, FOCUS, and MAGMA. This comprehensive integration ultimately resulted in the identification of two promising candidate genes, ATG4B and THAP4 (Supplementary Figure S4), that warrant further investigation as key regulators in sepsis. For the genomic locus 2:242149921–2:243188920, ATG4B demonstrated a posterior probability ranging from 0.933 to 1 across 22 tissues – including adipose subcutaneous, adrenal gland, artery aorta, brain cerebellar hemisphere, and colon sigmoid, among others (Supplementary Figure S5 A-3V) – while THAP4 achieved a posterior probability of 0.961 in the brain hippocampus (Supplementary Figure S5 W).
MR and colocalisation results
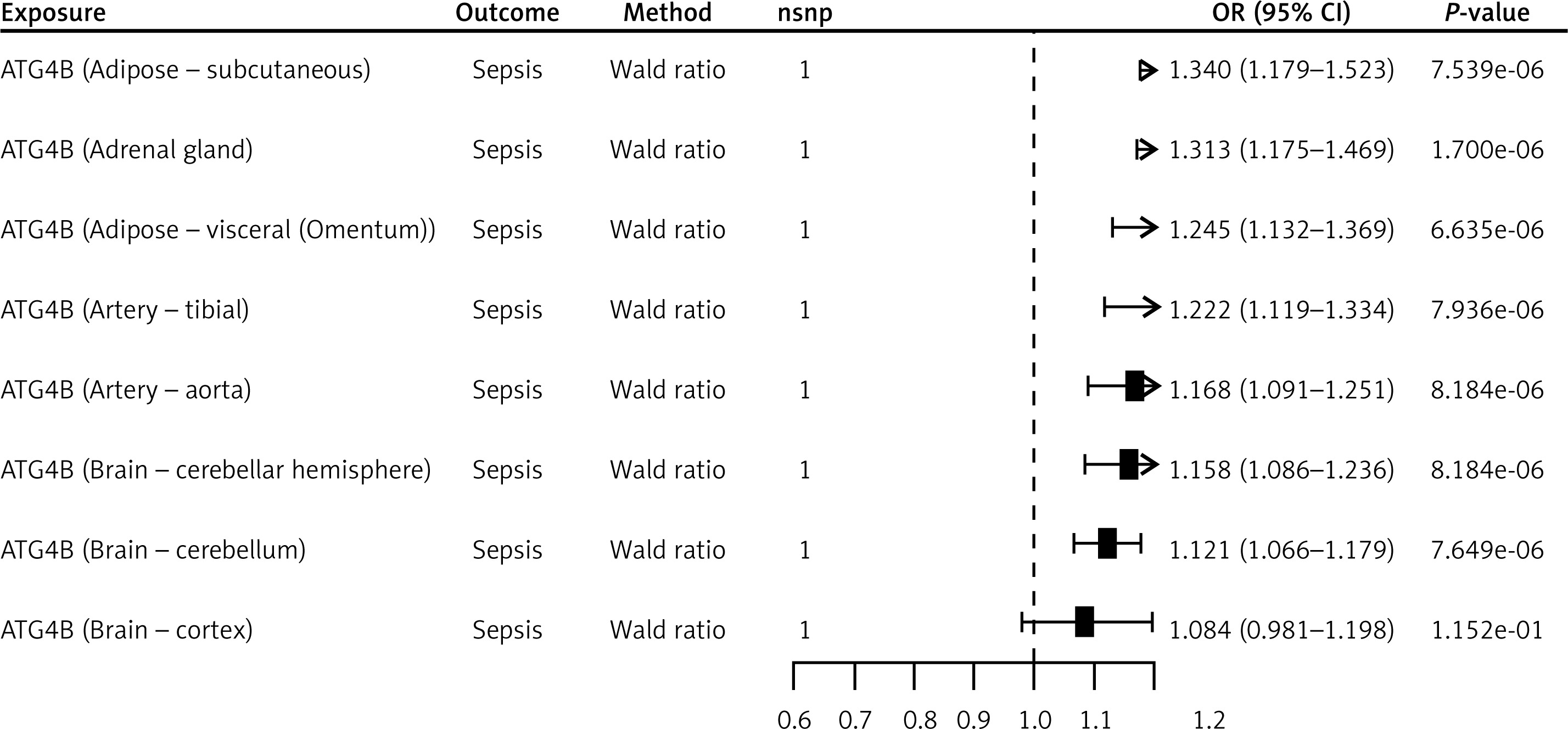
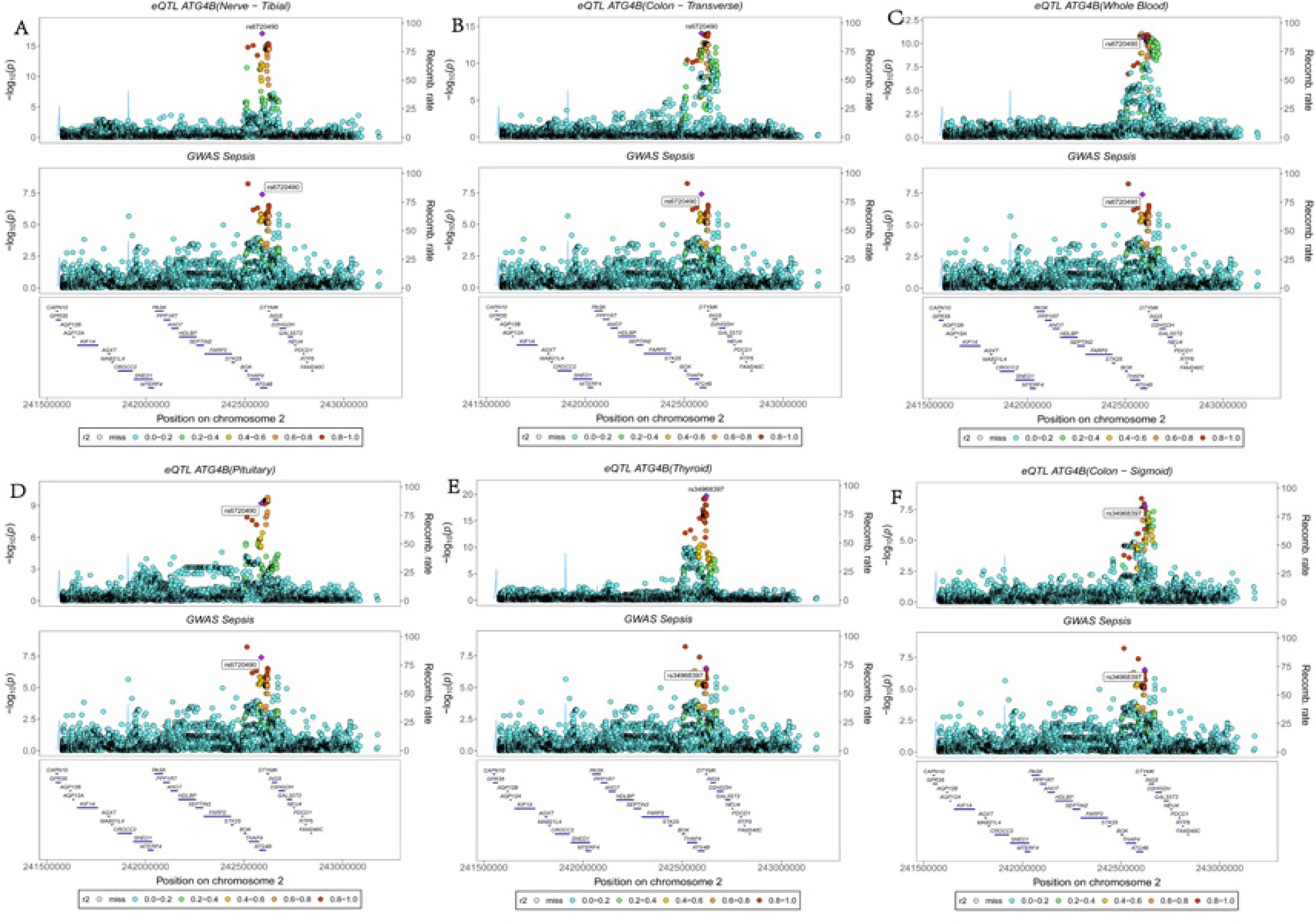
The ATG4B gene, located on chromosome 2q37.3, emerged as a key candidate in our multi-omics analyses. FUSION analysis demonstrated that ATG4B was significantly associated with sepsis across 27 tissues – including adipose subcutaneous, adrenal gland, brain cerebellum, lung, thyroid, uterus, and whole blood – highlighting its broad impact on the sepsis phenotype. MR analysis further supported this association by revealing significant effects in seven tissues (adipose subcutaneous, adrenal gland, adipose visceral (omentum), artery tibia, artery aorta, brain cerebellar hemisphere, and brain cerebellum), with odds ratios ranging from 1.12 to 1.34 (Figure 1). Complementary results were obtained via SMR analysis, which identified significant associations of ATG4B with sepsis in 14 tissues, including colon_ , nerve tibial, lung, thyroid, pituitary, and whole blood, with odds ratios between 1.12 and 1.37 (Figure 2, Supplementary Table SVIII). Subsequent colocalisation analysis provided further evidence of a causal relationship. ATG4B exhibited significant colocalisation effects in 6 tissues – including nerve tibial, colon transverse, whole blood, pituitary, thyroid and colon – sigmoid – with posterior probability of H4 (PP.H4) values ranging from 0.72 to 0.99 (Supplementary Table SVII). Notably, the single nucleotide polymorphism (SNP) rs6720490 emerged as the most significant colocalisation locus in tissues such as nerve tibial, colon transverse, whole blood, and pituitary (Figures 3 A–D). In parallel, rs34968397 was identified as the top colocalisation locus in thyroid and colon - sigmoid (Figures 3 E, F). Collectively, these results underscore the potential of ATG4B as a crucial regulator in sepsis pathogenesis and warrant further functional validation.
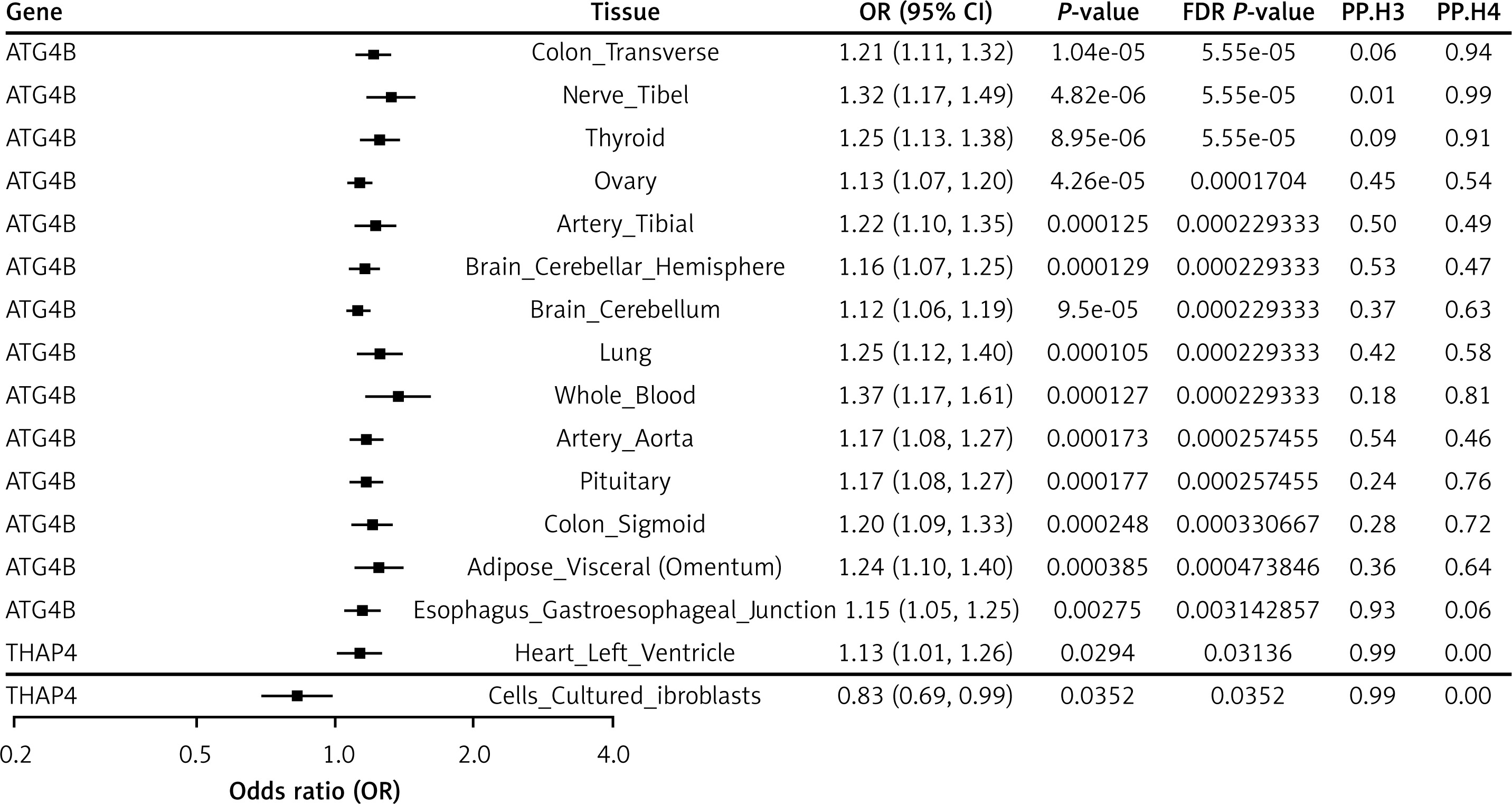
The THAP4 gene, also located on chromosome 2q37.3, exhibited significant expression in cells cultured fibroblasts, heart left ventricle, and stomach, according to the FUSION results. Due to insufficient instrumental variables, Mendelian randomisation analysis could not be conducted for THAP4, limiting our investigation to SMR analysis. Notably, the effect direction of THAP4 on sepsis risk was inconsistent across tissues. In heart left ventricle tissue, the SMR analysis yielded an odds ratio (OR) of 1.13 (95% CI: 1.01–1.26), suggesting a risk-enhancing effect, whereas in cells cultured fibroblasts the OR was 0.83 (95% CI: 0.69–0.99), indicating a protective effect (Figure 2, Supplementary Table SVIII). Furthermore, colocalisation analysis did not detect a significant colocalisation effect in any of these tissues (Supplementary Table SVII), which reduces the certainty of a causal relationship for THAP4 in sepsis.
Figure 3
The results of colocalisation analysis between ATG4B and sepsis. The SNP rs6720490 exhibited the lowest cumulative sum of sepsis GWAS and ATG4B eQTL p values in nerve tibial (A), colon transverse (B), whole blood (C), and pituitary (D). The SNP rs34968397 exhibited the lowest cumulative sum of sepsis GWAS and ATG4B eQTL p values both in thyroid (E) and colon sigmoid (F)
Discussion
Our study employed a comprehensive multi-omics approach that integrated GWAS, TWAS) (both cross-tissue and single-tissue), conditional and joint (COJO) analysis, gene-based testing using MAGMA, fine-mapping via FOCUS, MR, and SMR, and Bayesian colocalisation analysis. This integrative strategy allowed us to leverage diverse datasets and analytical methods to comprehensively assess the genetic architecture underlying sepsis. Through the cross-tissue and single-tissue TWAS analyses, we initially identified 2 candidate genes that exhibited robust associations with sepsis. These candidate genes were selected based on stringent statistical criteria across multiple tissues, underscoring their potential biological relevance in the pathogenesis of sepsis. The integration of results from UTMOST, FUSION, FOCUS, and MAGMA further refined our candidate gene set, ultimately pinpointing two particularly promising genes – ATG4B and THAP4 – as key regulators. ATG4B, in particular, demonstrated consistent and significant associations across numerous tissues along with strong supporting evidence from MR, SMR, and colocalisation analyses. In contrast, THAP4 exhibited significant expression in selected tissues but showed inconsistent effect directions and lacked robust colocalisation, suggesting that further investigation is warranted.
ATG4B is located on chromosome 2q37.3 and emerged as a noteworthy candidate based on its extensive and significant expression profile across multiple tissues, as demonstrated by our FUSION analysis. The gene was significantly associated with sepsis in as many as 27 tissues, including critical ones such as adipose subcutaneous, adrenal gland, brain cerebellum, lung, thyroid, uterus, and whole blood. This broad tissue expression underscores its potential widespread role in modulating the sepsis phenotype. Both MR and SMR analyses provided strong supportive evidence for the involvement of ATG4B in sepsis. MR analysis revealed significant associations in seven tissues (adipose subcutaneous, adrenal gland, adipose visceral, artery tibia, artery aorta, brain cerebellar hemisphere, and brain cerebellum), with odds ratios ranging from 1.12 to 1.34. Meanwhile, SMR analysis confirmed significant associations in 14 tissues, including colon transverse, nerve tibial, lung, thyroid, pituitary, and whole blood, with odds ratios between 1.12 and 1.37. The consistency of these findings across different tissues and methodologies bolsters the hypothesis that ATG4B plays a causal role in sepsis pathogenesis. Further supporting the causal role of ATG4B, our colocalisation analysis revealed significant overlap between sepsis GWAS and eQTL signals for this gene, with posterior probability of H4 (PP.H4) values ranging from 0.72 to 0.99 across 11 tissues, including nerve tibial, colon transverse, spleen, thyroid, brain – nucleus accumbens (basal ganglia), and whole blood. The PP.H4 values exceeding the threshold of 0.70 indicate a strong likelihood that the same genetic variants underlying the expression of ATG4B are also responsible for the sepsis phenotype, thereby providing robust statistical support for its causal relationship. rs6720490 was identified as the most significant colocalisation locus in tissues such as nerve tibial, colon transverse, whole blood, oesophagus mucosa, and pituitary. Its repeated appearance across multiple tissues underscores its potential impact on ATG4B expression and, by extension, sepsis susceptibility. rs56306534 was highlighted as a top colocalisation signal in spleen, brain – nucleus accumbens, breast – mammary tissue, muscle – skeletal, and pituitary, suggesting that this locus may influence ATG4B-mediated pathways in a tissue-specific manner. rs34968397 emerged as the most significant signal in thyroid and colon sigmoid, further validating the gene’s role and emphasising the regional specificity of its regulatory influence. Importantly, the prioritisation of ATG4B was not based solely on its cross-tissue TWAS significance. Instead, convergent evidence from fine-mapping, conditional analysis, Mendelian randomisation, SMR, and Bayesian colocalisation analyses collectively supports a putative causal role of ATG4B in sepsis susceptibility.
THAP4 is located on chromosome 2q37.3 and exhibits significant expression in a select set of tissues, as highlighted by our FUSION analysis. Notably, it demonstrated marked expression in cells cultured fibroblasts, heart left ventricle, and stomach, suggesting a potential tissue-specific role in influencing sepsis susceptibility. Unlike ATG4B, MR analysis for THAP4 could not be performed due to the lack of sufficient instrumental variables. This limitation restricts the direct inference of a causal relationship through MR, thereby necessitating reliance on alternative approaches such as SMR to evaluate its association with sepsis. The SMR analysis yielded conflicting effect directions for THAP4 across different tissues. In heart left ventricle tissue, the gene exhibited a risk-enhancing effect (OR = 1.13, 95% CI: 1.01–1.26), whereas in cells cultured fibroblasts it showed a protective effect (OR = 0.83, 95% CI: 0.69–0.99). These inconsistencies in the effect size and direction reduce the confidence in a uniform biological impact of THAP4 on sepsis risk across tissues. Furthermore, the colocalisation analysis did not reveal significant overlap between the sepsis GWAS and eQTL signals for THAP4 in the tissues examined. The lack of robust colocalisation evidence further diminishes our confidence regarding a causal role for THAP4 in sepsis pathogenesis.
Multi-omics analyses identify ATG4B as a key candidate in sepsis, with consistent associations across multiple tissues, suggesting a mechanistic role in autophagy, inflammation, and stress responses [12, 13]. ATG4B is involved in autophagosome formation, essential for cellular homeostasis and recycling damaged organelles. Dysregulation of ATG4B may impair autophagic flux, exacerbating inflammation and cellular stress during sepsis [14]. These findings highlight ATG4B as a potential therapeutic target. THAP4, a member of the THAP family, is implicated in transcriptional regulation and apoptosis [15]. While it shows significant expression in specific tissues, its inconsistent effects across tissues suggest a context-dependent role in sepsis. Further research into the molecular mechanisms of THAP4 is needed to determine its potential as a therapeutic target in sepsis.
Our study demonstrates the value of integrating analytical methods like UTMOST, FUSION, FOCUS, and MAGMA to explore sepsis genetics, improving candidate gene identification and reducing false positives. While ATG4B and THAP4 emerged as promising regulators, limitations such as insufficient MR instrumental variables, tissue sample size variations, and potential confounders need consideration. Further validation is required to confirm these findings and better understand the molecular mechanisms of sepsis pathogenesis.
In conclusion, our multi-omics approach suggests a potential role of ATG4B in autophagy, inflammation, and stress responses in sepsis, supported by consistent tissue associations and MR/SMR analyses. Although THAP4 showed tissue-specific expression, its inconsistent effects and limited colocalisation require further investigation. Additional experimental validation is needed to confirm these genes as therapeutic targets for sepsis.