Pulmonary hypertension (PH) is a severe, progressive disease which may be idiopathic or is associated with or is a sequela of underlying other disease. It is defined as elevation of mean pulmonary artery pressure (mPAP) of ≥ 25 mm Hg at cardiac catheterization. A clinical classification characterizes disorders causing PH into five groups, based on similar pathological findings, hemodynamic characteristics, and management (Table I) [1]. Pulmonary hypertension associated with left heart disease (PH-LHD) is classified as group 2 PH and represents the most prevalent form of PH [2].
Table I
Updated clinical classification of pulmonary hypertension [1]
Typically many patients with left heart disease develop PH as their primary cardiac disorder progresses. Hence the prevalence of PH increases in a particular left heart disease with advancing functional class (Table II) [3–8]. PH-LHD is common in patients with heart failure with preserved (HF-PEF) [4] or reduced ejection fraction (HF-REF) [9] and in valvular heart disease [5–8]. When present, PH-LHD results in more severe symptoms and worse exercise tolerance [10–13]. In general the development of PH in a patient with left heart disease portends a poor prognosis vis-a-vis normal pulmonary artery pressures [14].
Table II
Prevalence of pulmonary hypertension in left heart disease
| S. No. | Left sided heart disease | Prevalence of PH (%) |
|---|---|---|
| 1 | LV systolic dysfunction [3] | 60 |
| 2 | LV diastolic dysfunction (PASP > 35 mm Hg) [4] | 83 |
| 3 | Mitral stenosis (PASP ≥ 50 mm Hg) [5] | 38 |
| 4 | Mitral regurgitation (PASP > 50 mm Hg) [6] | 23 |
| 5 | Aortic stenosis (PASP > 50 mm Hg) [7] | 29 |
| 6 | Aortic regurgitation (PASP ≥ 60 mm Hg) [8] | 16 |
[i] Note: The above percentages are only approximate values based on the quoted series. It must be emphasized that the occurrence of PH is dependent on severity and duration of left heart disease such that the PH is more common in patients in advanced stages of underlying left heart disease. Further, it is not necessary that all patients with similar severity of particular left heart disease will develop PH or if PH is present it is of equal severity. Nevertheless, the presence and severity of PH signifies poor prognosis. PASP – pulmonary arterial systolic pressure, LV – left ventricular.
Pre-capillary and post-capillary pulmonary hypertension
If the underlying pathology is predominantly in the pulmonary arterioles and small pulmonary arteries, PH is termed pre-capillary. If the underlying cause of raised PAP is left heart disease, PH is termed isolated post-capillary PH (Ipc-PH). By definition, in Ipc-PH the raised PAP is a passive phenomenon meaning there is no intrinsic pathology in pulmonary circulation (at least in the beginning) so as to maintain forward flow in response to elevated left sided filling pressures. However in many patients chronic passive elevation of PAP leads to pathologic changes in the small pulmonary arteries and arterioles such that the process is no longer passive. The raised PAP in such cases has dual cause: elevated left sided filling pressures and intrinsic pulmonary vascular disease termed combined post-capillary and pre-capillary PH (Cpc-PH).
Hemodynamics
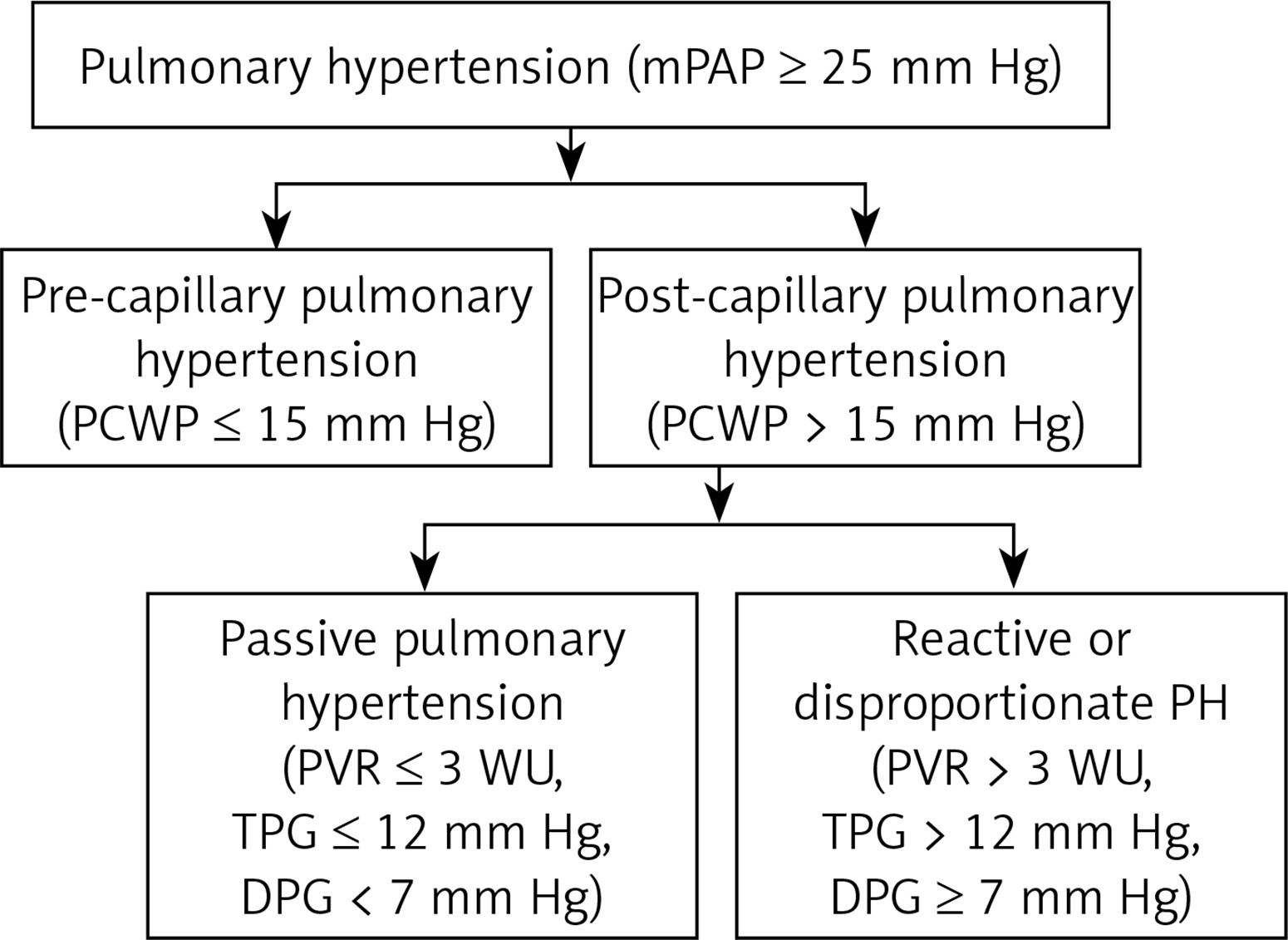
Pulmonary hypertension is defined hemodynamically by invasive right heart catheterization (RHC) as mPAP ≥ 25 mm Hg and a normal or reduced cardiac output. The severity of PH is graded by Doppler echocardiography based on estimated peak PAP as mild 36–45 mm Hg, moderate 46–60 mm Hg and severe if > 60 mm Hg. The RHC based grading is by mean PAP: mild 25–40 mm Hg, moderate 41–55 mm Hg and severe > 55 mm Hg [12, 15–17]. The main hemodynamic parameter that differentiates PH-LHD is pulmonary capillary wedge pressure (PCWP) measured at end-expiration. A PCWP cut-off of 15 mm Hg defines whether PH is pre-capillary (PCWP ≤ 15 mm Hg) or post-capillary (PCWP > 15 mm Hg).
Transpulmonary gradient (TPG) and diastolic pulmonary gradient (DPG) differentiate whether PH is passive or reactive. The TPG is defined as difference between mean PAP and mean PCWP (TPG = mPAP – mPCWP). A TPG < 12 mm Hg defines passive PH and TPG ≥ 12 mm Hg defines reactive PH. Diastolic PAP is less influenced by increased PCWP than systolic PAP and mean PAP. Therefore, raised PCWP will result in marked increase in systolic PAP and mean PAP with no or little increase in diastolic PAP [13]. Diastolic pulmonary gradient defined as difference between diastolic pulmonary artery pressure (dPAP) and mean PCWP (DPG = dPAP – mPCWP) is a marker of pulmonary vascular disease. In normal subjects DPG is 1–3 mm Hg. The raised diastolic pulmonary gradient (> 7 mm Hg) in a given patient of PH-LHD suggests setting-in of pre-capillary component (pulmonary vascular pathology) over and above post-capillary component [13]. In one study the patients with TPG > 12 mm Hg who had DPG > 7 mm Hg had worse prognosis [18]. In contrast, another study of 1236 patients with cardiomyopathy and PH-LHD, elevated DPG was not associated with worse survival [19]. Pulmonary vascular resistance (PVR) of ≤ 3 Wood units (WU) characterizes Ipc-PH, whereas PVR > 3 WU is suggestive of Cpc-PH or reactive PH in the setting of left heart disease (Figure 1). The latest ESC/ERS guidelines recommend combined use of DPG and PVR to differentiate between Ipc-PH and Cpc-PH in PH-LHD [20].
Figure 1
Hemodynamic definition of pulmonary hypertension
TPG = mPAP – mPCWP, DPG = dPAP – mPCWP, DPG – diastolic pulmonary gradient, dPAP – diastolic pulmonary artery pressure, TPG – transpulmonary gradient, mPAP – mean pulmonary arterial pressure, mPCWP – mean pulmonary capillary wedge pressure, PH – pulmonary hypertension, PVR – pulmonary vascular resistance, WU – Wood units.
Impact of pulmonary hypertension development on prognosis
The development of PH in left heart disease is a marker of chronicity. Patients with PH-LHD typically have advanced disease and have poorer prognosis [11, 21]. Pulmonary hypertension is associated with decreased survival in HF-PEF and HF-REF [4, 9]. In patients with PH-LHD, the right ventricular (RV) function further dichotomizes the risk. The presence of RV dysfunction in patients with PH-LHD is an ominous finding. Ghio et al. have reported RV dysfunction with high mPAP in 57% of their cohort of patients with dilated cardiomyopathy [21]. They found a significant inverse relationship between mPAP and RV ejection fraction. Nonetheless many patients of dilated cardiomyopathy with normal mPAP had RV dysfunction. The presence of RV dysfunction gives prognostic information over and above provided by mPAP [21].
Pre-transplant PH is associated with higher early mortality and poor post-transplant survival. Therefore, assessment of severity of PH is important in determining the candidacy for heart transplantation (HT). In patients with PASP ≥ 50 mm Hg and either TPG ≥ 15 mm Hg or PVR > 3 WU, vasodilator challenge should be administered to see for reversibility of PH. Patients showing reversibility have better prognosis than patients with fixed PH. If initial acute vasodilator challenge is unsuccessful, medical management with diuretics, nitric oxide and inotropes and/or mechanical assist devices may be tried in an attempt to decrease PVR. If PVR fails to decrease by above measures, patient is deemed to have irreversible fixed PH [22]. Severe fixed PH is a contraindication to HT. Oral sildenafil can be tried in patients with irreversible PH associated with heart failure in an attempt to decrease PVR and as a bridge to transplantation. In a study of 18 patients with severe LV dysfunction, sildenafil increased RV ejection fraction from 26 ±7% to 30 ±9%, p = 0.008 and decreased PVR from 5.3 ±1.9 to 3.3 ±1.8 WU, p = 0.01 over a median follow-up of 8.7 months. During follow-up, 5 patients had HT and 6 had LV assist device implantation. All patients showing significant response as evidenced by a decrease in PVR to < 3 WU were alive at follow up compared to non responders who had 44% survival rate [23]. In a comparative study of 15 patients with severe PH receiving oral sildenafil therapy and 104 patients without severe PH who underwent heart transplantation, sildenafil was administered in severe PH group for 163 ±116 days before HT, and 43 ±45 days after HT. Sildenafil therapy resulted in significant decrease in PVR from 5.0 ±1.1 to 3.0 ±1.6 WU, mean TPG decreased from 17.3 ±3.2 to 10.2 ±4.1 mm Hg, and mPAP decreased from 43.9 ±12.5 to 33.4 ±5.8 mm Hg, p < 0.01. As a consequence all severe PH patients could undergo successful HT. Sildenafil could be withdrawn in most patients late after HT. Survival after HT was similar between both the groups [24].
Pulmonary hypertension and RV dysfunction after HT is associated with 3–5 fold increased risk of 30 day mortality. About 10–15% of all HT recipients develop RV dysfunction after transplantation. The addition of oral sildenafil in the post-operative period may allow early withdrawal of inhaled nitric oxide and intravenous drugs administered to reduce PAP [25].
Persistent/residual pulmonary hypertension in valvular heart disease
Most patients of valvular heart disease will have significant decrease in PAP after relief of valve stenosis/regurgitation [26, 27]. However, persistent/residual PH is observed in many patients even after successful relief of mitral valve obstruction or regurgitation by percutaneous balloon mitral valvotomy (BMV) or surgical mitral valve replacement [26]. Besides the presence of fixed component of PH, other factors like suboptimal BMV, significant MR after BMV and patient-prosthesis mismatch are responsible for the persistence of PH even after therapeutic intervention [28–30].
In the study by Puri et al. residual PH correlated with functional capacity impairment 7 days after mitral valve replacement as determined by 6 min walk test [31]. Another study compared 287 patients with persistent PH 1 year after BMV with 414 patients who did not have PH after BMV. The patients with persistent PH were older, had severe pre-BMV PH, increased Wilkin’s mitral valve echocardiography scores and suboptimal BMV result. Importantly, on long term follow-up persistent PH group had more new onset heart failure and higher need for reintervention [32]. Further patients with atrial fibrillation have increased right atrial dilatation which further increases tricuspid annular dilatation and tricuspid regurgitation [33].
Pathophysiology of pulmonary hypertension in left heart disease
The increased pulmonary venous pressure results in disruption of alveolar-capillary walls termed alveolar-capillary stress failure, resulting in capillary leakage and acute alveolar edema [34]. This acute stage is reversible. However with chronically increased pulmonary venous pressure there is irreversible remodeling of the alveolar-capillary membrane as a compensatory mechanism to decrease the frequency and severity of potentially life-threatening pulmonary edema [35].
The remodeling affects both pulmonary venous and arterial system with thickening of the capillary endothelial and alveolar epithelial cell basement membranes and pulmonary veins. These changes reduce the permeability of the alveolar-capillary membrane to fluids, and prevent development of pulmonary edema. The process also results in muscularization of the arterioles and neointima formation along with medial hypertrophy of distal small pulmonary arteries leading to increased pulmonary vascular resistance [36, 37]. Also lymphatic vessels are dilated [35]. With long-standing disease, pulmonary edema becomes less frequent and the clinical picture is dominated by development of PH and right heart failure (Figure 2).
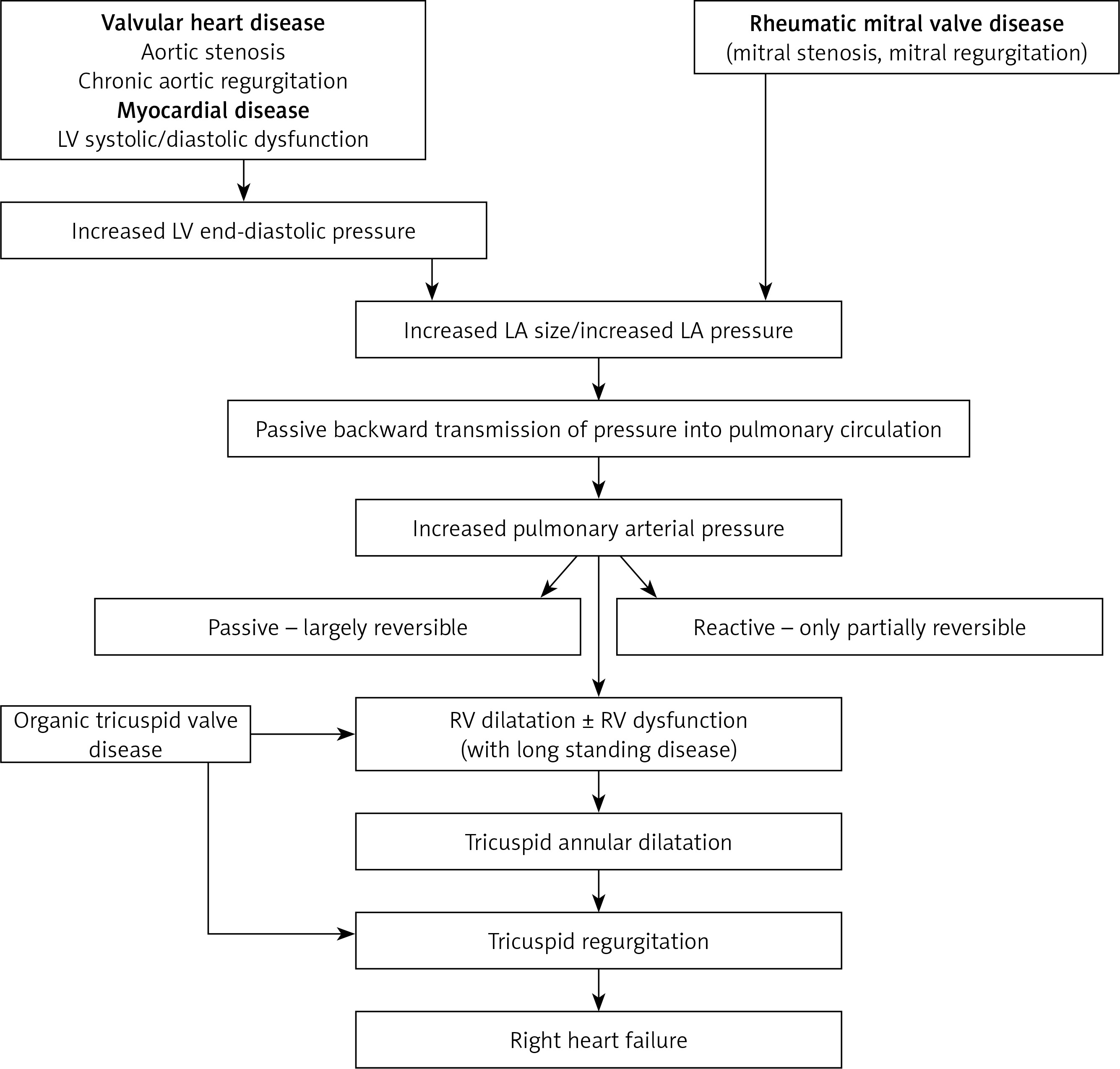
The development of pulmonary vascular disease is variable, with some patients developing severe PH while others being spared of PH despite similar rises in PCWP. While why this happens is unknown, some factors may be responsible. The patients with large compliant left atria may be less prone to development of pulmonary edema and ultimately less severe PH. Also development of AF may make them more prone to develop PH.
Pulmonary function abnormalities
The gas diffusion across the alveolar capillary membrane is decreased in HF, the degree of impairment depending on severity of HF [38]. Patients with mitral valve disease have decreased FVC, FEV1 and diffusion capacity of lung for carbon monoxide (DLCO) and increase in residual volume [39]. Structural changes in the alveolar-capillary membrane decrease diffusion capacity of the lung with resultant impedance to gas transfer contributing to exercise intolerance.
Genetics
The influence of genetics in development of idiopathic PH (WHO Group 1) is undisputed. Genetic studies have shown that approximately 80% of patients with familial pulmonary arterial hypertension (PAH) and 25% of patients with sporadic PAH carry mutations in the bone morphogenetic protein receptor 2 (BMPR II) [1]. But there are no studies which have investigated a link between PH-LHD and BMPR II mutations. Endothelin-1 gene polymorphisms have been shown to induce pulmonary hypertension in idiopathic PAH [40, 41]. Although there is no concrete evidence for genetic predisposition, attempt is being made to find predisposing genetic factors in PH-LHD. Serotonin and its transporter protein (5-HTT) have vasoactive and mitogenic properties. Homozygous patients for the long variant of 5-HTT have been shown to be associated with elevated PAP in heart failure [42].
Two-dimensional and Doppler echocardiography
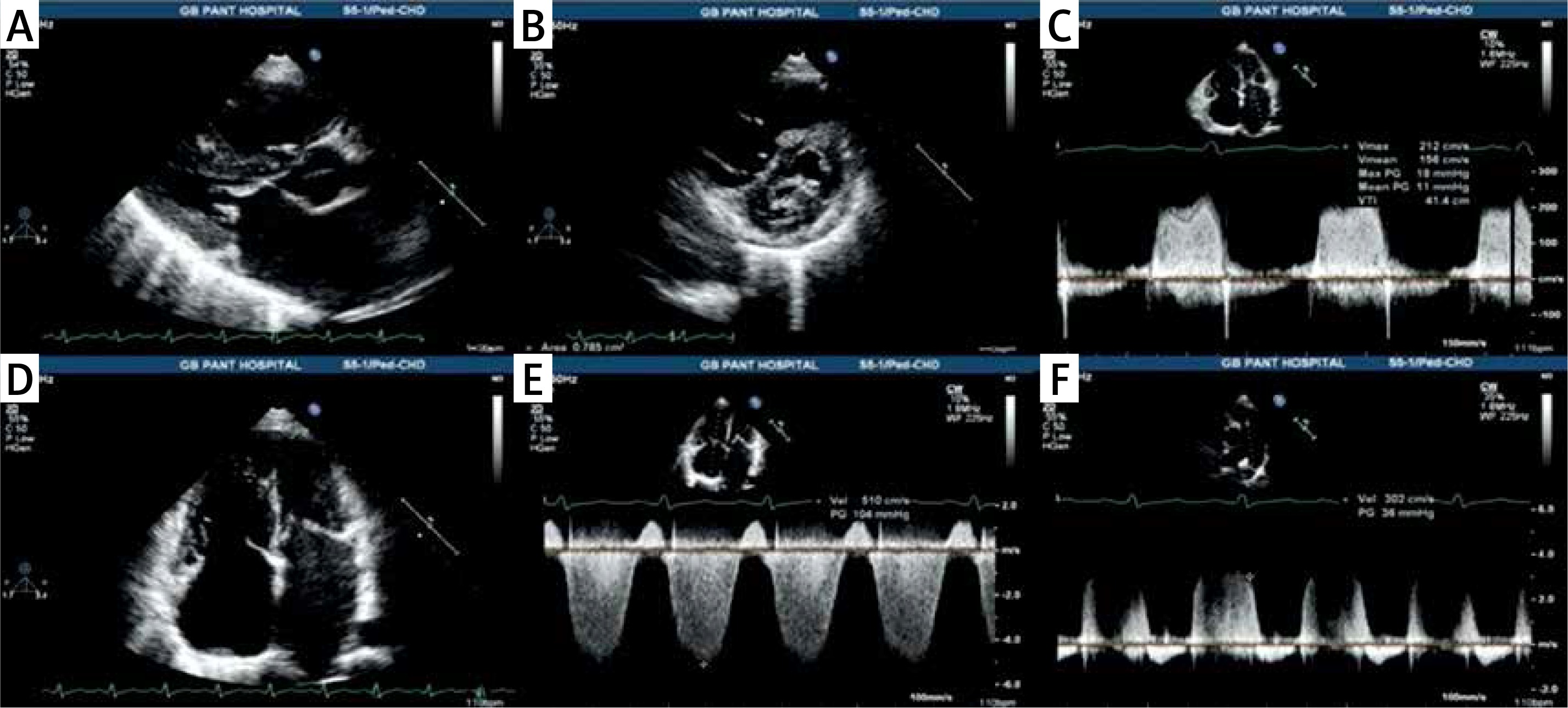
Echocardiography is used for the diagnosis and quantification of severity of left heart disease like left ventricular systolic/diastolic dysfunction and valvular heart disease and highlights features suggestive of PH like right atrial (RA) enlargement and RV dilatation, hypertrophy or dysfunction. Echocardiography is a semi-quantitative screening tool for diagnosing PH [43–47]. Doppler echocardiography is used to estimate the right ventricular systolic pressure (RVSP) from tricuspid regurgitation velocity jet by adding estimated RA pressure. The ancillary signs of RV involvement must be present in addition to elevated RVSP to diagnose PH (Table III) [48]. Also peak early diastolic and end diastolic velocities of pulmonary regurgitation (PR) jet continuous wave Doppler trace significantly correlate with mean and end diastolic PAP respectively [49] (Figure 3).
Table III
Figure 3
Echocardiography of patient in Fig. 3 of rheumatic heart disease with severe mitral stenosis in normal sinus rhythm showing: A – thickened mitral leaflets with diastolic doming of anterior mitral leaflet with marked left atrial dilatation, B – mitral valve in short-axis in diastole showing fused medial and lateral commissures with area of 0.78 cm2 by planimetry, C – measurement of mean transmitral gradient of 11 mm Hg, D – dilated left atrium, right atrium and right ventricle, E – CW Doppler of TR jet showing peak velocity of 5.1 m/s with calculated RVSP of 104 mm Hg plus right atrial pressure, F – end-diastolic velocity of pulmonary regurgitation signal of 3 m/s with calculated pulmonary artery end diastolic pressure of 36 mm Hg plus right atrial pressure
The grading of severity of PH is ideally done by RHC especially when specific pulmonary vasodilator therapy is planned or in the event of diagnostic ambiguity. Although only modest correlation of Doppler derived PA pressures and RHC measured pressures has been reported in earlier studies (r ≈ 0.7) [50], a recent study of 667 patients undergoing both RHC and transthoracic echocardiography has reported a high correlation of invasive mean PAP and Doppler derived systolic PAP in patients with interpretable TR jets. The degree of correlation decreased in patients who had partially visible TR jet envelope, signifying the importance of TR jet envelope quality in correctly estimating systolic PAP [51]. Echocardiographic features suggesting post-capillary PH include larger left heart chambers compared to right heart chambers with LV forming the apex, LV eccentricity index < 1.2 and E/E’ ≥ 10 [52]. The Doppler derived systolic PAP can be monitored periodically non-invasively along with RV assessment by echocardiography.
However, the degree of improvement in RV size, septal position and RV systolic function measured by tricuspid annulus plane systolic excursion (TAPSE), rather than Doppler-estimated PASP in response to PH-specific therapy are predictive of outcomes in Group 1 PH [53]. TAPSE has also been shown to assist in risk stratification in acute pulmonary embolism [54]. Although not specifically studied in PH-LHD, it may be reasonable to follow these patients with regards to above parameters. Other parameters like PA elastance can be calculated by echocardiography which shows moderate correlation with RHC-derived PVR [55].
Right heart catheterization
Right heart catheterization is the gold standard for diagnosis of PH and is recommended in patients with PH-LHD [20, 56]. When reliable PCWP cannot be measured on RHC, left heart catheterization is recommended. Right heart catheterization can also assist in ruling out other causes of PH [3, 20]. Exercise testing may be helpful in evaluation of patients with borderline PH associated with left heart disease [56]. Further, fluid challenge with fluid load of 500 ml can unmask underlying LV diastolic dysfunction and occult pulmonary venous hypertension by documenting increased LVEDP to > 15 mm Hg in patients where baseline values were normal [57].
The presence and quantification of PH may explain patient symptoms, affect management decisions and help in assessing prognosis. Resting PAP > 50 mm Hg is Class IIa indication for intervention in asymptomatic severe mitral stenosis (ESC guidelines) [58] and asymptomatic severe mitral regurgitation (MR) (ESC and AHA guidelines) [58, 59]. Exercise induced PH in asymptomatic patients with valvular heart disease and in patients with secondary MR may provide additional prognostic information (Table IV) [60–73].
Table IV
Role of exercise in evaluation of valvular disease
| Role of exercise in mitral stenosis | Role of exercise in aortic stenosis | Role of exercise in primary mitral regurgitation (MR) | Role of exercise in secondary mitral regurgitation |
|---|---|---|---|
|
|
|
|
[i] AS – aortic stenosis, CHF – chronic congestive heart failure, ERO – effective regurgitant orifice, LA – left atrium, LV – left ventricle, LVEF – left ventricular ejection fraction, MS – mitral stenosis, MR – mitral regurgitation, PAP – pulmonary artery pressure, PH – pulmonary hypertension, RV – right ventricle.
Management
The management of PH-LHD predominantly involves the treatment of underlying left heart disease. Aggressive medical management of HF-REF primarily includes diuretics, angiotensin converting enzyme inhibitors, angiotensin receptor blockers, β-blockers and mineralocorticoid receptor antagonists [74]. Digoxin can be used as second line drug in patients with persisting symptoms despite being on above mentioned drugs [74]. Corrective valve surgery/percutaneous intervention is recommended and is usually associated with resolution of PH [59]. The recent approval of new drugs for pulmonary artery hypertension has rekindled the interest in the treatment of PH. However the new drugs introduced are for WHO categories Group 1 PH and Group 4 PH. Unfortunately despite being the commonest cause of raised pulmonary artery pressures, PH-LHD remains an under-studied disease with no specific treatment available. Despite promising results in acute hemodynamic studies with some pulmonary vasodilators, the intermediate term results have been disappointing. The reasons for this are many and include: 1) the non-specific selection of patients with systolic and diastolic dysfunction in most studies without adequate segregation based on baseline pulmonary artery pressures except in few studies [75, 76], 2) no studies are available on PH associated with valvular heart disease, an important subset of PH-LHD. At present, there is no evidence that PH specific drug therapy is effective in patients with PH-LHD. Further, the European Society of Cardiology/European Respiratory Society guidelines do not recommend the use of drugs approved for Group 1 PH in PH-LHD due to the lack of evidence [3].
Nitric oxide (NO) and prostacyclin are potent pulmonary vasodilators which improve pulmonary hemodynamics by decreasing pulmonary vascular resistance [77]. The endothelin system is activated in chronic heart failure and elevated plasma ET-1 levels correlate with hemodynamic severity of heart failure and correlate with mortality [78]. Plasma ET-1 acts via two receptor subtypes (ETA and ETB) which leads to pulmonary vasoconstriction, and vascular smooth muscle cell proliferation. In small studies using the dual endothelin-1 receptor antagonist bosentan and tezosentan, and selective endothelin A receptor antagonist darusentan, there was an acute improvement in mean PAP, right atrial pressure, PCWP, and cardiac output [78–81]. Intravenous bosentan led to acute improvement in pulmonary hemodynamics in 24 patients with chronic heart failure [78]. In a large placebo-controlled study of patients with acute decompensated heart failure, intravenous tezosentan led to an acute reduction in left ventricular filling pressures, and increased cardiac index but did not result in improvement in dyspnea or pulmonary edema endpoints [78, 82–84]. Further in another large study of 1435 acute HF patients with persistent dyspnea, the dyspnea score, incidence of death and worsening HF was similar between intravenous tezosentan for 24–72 h group and placebo [85]. A number of longer-term studies have not confirmed a benefit for the use of endothelin receptor antagonists in patients with PH associated with HF. However, there was a trend toward reduced HF mortality and morbidity with bosentan [86]. Currently the use of endothelin receptor antagonists is not recommended for the treatment of PH associated with left heart disease.
It is likely that the patients who have PH disproportionate to their underlying left heart disease will likely benefit from specific PH therapy. However, many of the trials included all patients with congestive heart failure, rather than patients specifically with associated PH. Only few small studies have studied effect of sildenafil in HF patients with PH [87, 88]. Although hypothetically pulmonary vasodilatation may result in increased cardiac filling resulting in increased pulmonary venous congestion, acute hemodynamic study by Lepore et al. showed that PDE-5 inhibition with sildenafil improves cardiac output by balanced pulmonary and systemic vasodilatation [88]. Data from FIRST study suggests that epoprostenol use in systolic heart failure is associated with increased mortality despite decrease in PVR and PCWP and increased cardiac output [89]. Similarly there was increased risk of worsening CHF with endothelin antagonist bosentan in systolic HF initially [86]. However patients who continued treatment for 6 months there was improvement in primary end-point [86]. Perhaps aggressive diuretic therapy before initiation of pulmonary vasodilator therapy and use of drugs which result in both pulmonary and systemic vasodilatation will be beneficial.
PDE-5 inhibitors such as sildenafil lead to pulmonary vasodilatation and have anti-proliferative action on pulmonary vascular smooth muscle cells. In chronic heart failure, sildenafil acutely lowers PVR and pulmonary arterial pressures, and improves endothelium dependent flow-mediated pulmonary vasodilatation, with a sustained clinical benefit at 6 months [75, 87, 88, 90]. In CHF with PH, sildenafil is associated with increased 6MWT distance, and improved pulmonary hemodynamics at 12 weeks [87, 88]. Longer-term studies have shown that sildenafil is associated with improvement in exercise capacity, cardiac output during and skeletal muscle blood flow during exercise [87]. However, sildenafil in HF-PEF did not result in any significant improvement in exercise capacity or clinical status over 24 weeks [76]. Despite these promising results, the routine use of sildenafil in patients with PH associated with left heart disease cannot be recommended.
Riociguat is a novel soluble guanylate cyclase (sGC) stimulator that sensitizes sGC to endogenous NO and directly stimulates sGC independently of NO, thereby increasing NO by dual means. In addition to causing vasodilatation, it has antifibrotic, anti-proliferative, and anti-inflammatory effects [91]. Although approved for Group 1 and 4 PH, a recent study of 201 patients having PH due to systolic heart failure riociguat did not result in any significant change in mean PAP, biomarkers, and CV death or hospitalization versus placebo despite improvement in stroke volume and cardiac index and decrease in pulmonary vascular resistance and systemic vascular resistance [92].
Conclusions
Pulmonary hypertension is common in patients with left heart disease, and is associated with increased morbidity and mortality. Pathophysiologic mechanisms are complex and comprise of both passive and active components secondary to left atrial hypertension. Echocardiography is screening tool for initial evaluation of patients with PH-LHD. Management of PH focuses upon the treatment of the underlying left heart disease, and associated comorbidities. Although there is no evidence for routine use of PH specific therapies in PH-LHD, there is some suggestion that PDE-5 inhibitors may be useful especially in certain subsets.
Future directions
At present management of PH-LHD is far from satisfactory. No clear cut guidelines exist as how to manage and follow-up PH-LHD patients. As some patients with similar left heart disease develop PH, while others do not, the factors which predispose to the development of PH need to be elucidated. As genetic predisposition is proven in idiopathic pulmonary arterial hypertension, the role of genetics in PH-LHD needs to be studied. There is a need to better define the echocardiographic markers to differentiate between Ipc-PH and Cpc-PH. As repeated catheterization studies to see the effect of management strategies may be impractical, better echocardiographic prognostic parameters need to be defined. The HF-PEF and HF-REF patients with PH need to be studied separately. Studies of pulmonary vasodilators need to dichotomize patients according to PAP rather than non-specific inclusion of a particular left heart disease. Trials of pulmonary vasodilators in PH associated with valvular heart disease need to be conducted. Future studies targeting lung structural and vascular remodeling in PH rather than only vasodilators will be necessary [93].