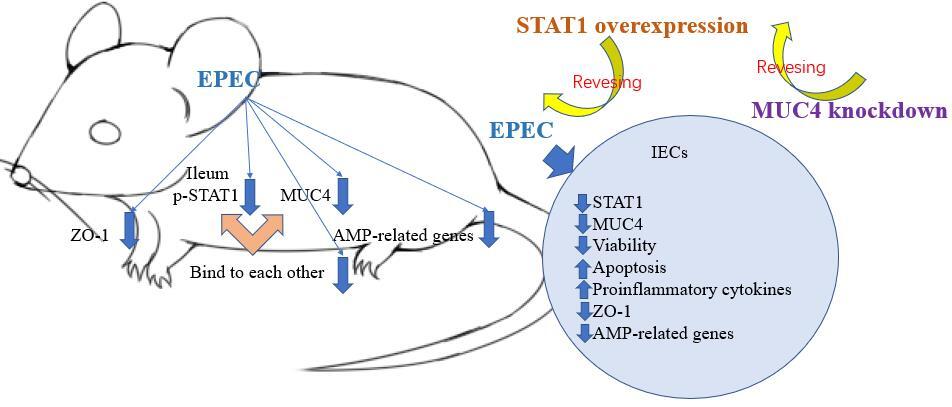
Introduction
Escherichia coli is a Gram-negative rod-shaped bacterium that has a commensal relation with humans without exerting any adverse effects [1]. However, commensal E. coli can be pathogenic, once it transforms into pathovars such asenteropathogenic E. coli (EPEC), which colonize the gastrointestinal (GI) tract, causing diarrheagenic and extraintestinal diseases in humans [2]. Additionally, EPEC is usually present in the food and water, and therefore is also known as a food-borne pathogen [3]. EPEC infection also induces malnutrition, which further increases the severity of the diseases caused by EPEC [4, 5]. It has been reported that EPEC infection is associated with remarkable morbidity and mortality among people with diarrhea in developing countries [6].
The pathogenic mechanism of EPEC involves interaction with enterocytes to form attaching and effacing (A/E) lesions that give rise to destruction of the microvilli, actin pedestal formation, and reorganization of the cytoskeleton, followed by reduction of the epithelial barrier area [7, 8]. Tight junctions, which reside at the apical part of the lateral membrane of the intestinal epithelial barrier, are assembled from cytoplasmic scaffolding proteins such as zonula occludens-1 (ZO-1), a scaffolding membrane protein [9]. Tight junctions serve to connect intestinal epithelial cells, maintain barrier polarity and regulate barrier function [10, 11]. Redistribution of tight junctions can be caused by EPEC-induced epithelial inflammation [12, 13], leading to altered barrier function.
Antimicrobial peptides (AMPs) are highly diverse and dynamic anti-infective molecules expressed by specific intestinal epithelial cells, Paneth cells, and immune cells in the GI tract [14, 15]. AMPs can also target virulence proteins [16]. A previous study showed that human α-defense in 5 (HD5), which is the most abundant Paneth cell-derived AMP, and its treatment of E. coli can cause blister morphology, which is associated with cell death [17]. Also, AMPs exhibit activity against a broad spectrum of bacteria through either affecting membrane stability or interfering with DNA replication, transcription, translation, protein biosynthesis, protein folding, or cell division [18–20]. Moreover, AMPs function as host immune response-modulating peptides to regulate gut microbial growth and composition [21]. Reduction of intestinal AMPs is associated with enteric dysbiosis and damage [22]. Research has shown that AMPs exert therapeutic effects against intestinal epithelial barrier inflammation and dysfunction caused by EPEC infection [23]. However, the upstream mechanism of the therapeutic effects has yet to be uncovered.
Previous research revealed that MUC4 deletion resulted in significant down-regulation of these AMPs at the mRNA level [24]. Also, stimulation of signal transducer and activator of transcription (STAT) 1 (STAT1) expression can induce MUC4 expression [25]. Furthermore, downregulation of STAT-1 has been recorded to be accompanied by a reduction in AMP formation, leading to intestinal microenvironmental disorders [22]. STAT1 is a member of intestinal STATs, which are a class of transcription factors crucial for transmitting intracellular signaling involved in cellular growth, differentiation, apoptosis, inflammation and immune responses [26, 27]. Evidence has indicated that STATs play essential roles in modulating the antimicrobial responses [28, 29]. Deficiency of STAT1 increases the susceptibility of mice to infection caused by microbial bacterial pathogens and viruses [30]. In addition, STAT1 has been documented to positively regulate the expression of mucin 4 (MUC4) by acting as a transcription factor of MUC4 [31]. MUC4 belongs to the transmembrane mucin family, whose secretion deters microorganisms [32]. MUC4 is normally expressed in the epithelia of several organs including the colon [33], where it can protect the apical surfaces of epithelial cells [34]. Additionally, MUC4 acts as an intramembrane ligand that modulates epithelial cell proliferation or differentiation after binding to ErbB2 [35]. Notably, knockout of MUC4 causes less distribution of AMPs in mice bearing colorectal cancer (CRC) [24]. Taking the evidence together, it is hypothesized that STAT1 upregulates the MUC4 level to induce AMP formation and thereby alleviate intestinal epithelial barrier injury caused by EPEC.
To test the hypothesis, the expression of and interaction between STAT1 and MUC4 were examined using in vivo models of EPEC-induced intestinal infections, and related in vitro models were established to confirm the role of the STAT1/MUC4 pathway in EPEC-caused intestinal epithelial barrier injury.
Material and methods
Animals
Male C57BL/6 mice (n = 18, aged 22 days, weighing 10–12 g) were housed in animal cages at 22 ±1.0°C, 50 ±5% humidity, with a 14-hour (h) light and 10-h dark circadian cycle. A standard rodent house chow diet was given to the mice from their arrival through the infection.
Preparation and culture of EPEC inocula
The bacterial strain used in the present study was EPEC E2348/69 (serotype O127:H6), which belongs to phylogroup B2 with its full-length chromosomal nucleotide accessible by the accession number: FM180568 [36]. Culture of EPEC was conducted in Dulbecco’s modified Eagle’s medium (DMEM; 11965092, Thermo Fisher, Waltham, MA, USA) at 37°C in a shaking incubator, and was terminated when the EPEC cultures turned orange, indicating optimal growth, with optical density (OD)600~0.6. The EPEC cultures underwent centrifugation at 3500 × g for 10 min (min) at 4°C to obtain bacterial pellets, which were then resuspended in DMEM to reach a density of 1010 CFU/ml [37].
Establishment of mouse models of EPEC infection
Mice were given drinking water containing an antibiotic cocktail (35 mg/l gentamicin (A1720, Sigma-Aldrich, St. Louis, MO, USA), 45 mg/l vancomycin (SBR00001, Sigma-Aldrich, USA), 215 mg/l metronidazole (M1547, Sigma-Aldrich, USA), and 0.085 mg/l colistin (HY-113678, MedChemExpress, Monmouth Junction, NJ, USA) for 3 days to disrupt resident microbiota; the mice showed hunching posture and diarrhea. Then, the antibiotics were cleared by drinking normal water for 1 day, after which infection with EPEC was carried out [38]. In brief, mice were randomized into two groups. Those (n = 6) in the EPEC group were orally challenged with 100 µl of DMEM containing 1010 CFU/ml EPEC via a 22-gauge feeding needle [37], while control mice (n = 6) were administered 100 µl of DMEM instead.
Three days after the infection [37], all the mice were anesthetized with 1% pentobarbital sodium (P010, 50 mg/kg, Sigma-Aldrich, USA) and sacrificed via decapitation. Mouse ileum was collected and used for molecular assays.
Immunohistochemical test
Mouse ileum was fixed with 4% paraformaldehyde (P885233, MACKLIN, Shanghai, China) for 24 h, followed by treatment with an ethanol and xylene gradient (95682, Sigma-Aldrich, USA). Then, the ileum was paraffinized (1496904, Sigma-Aldrich, USA) and cut into 4-µm-thick slices using a cryomicrotome (Leica CM 1850 UV, Leic Biosystems, Nussloch GmbH, Germany). After being deparaffinized, the slices were rehydrated and then water-boiled at 95°C with repair solution (P0088, Beyotime, Shanghai, China) for 10 min to repair antigen. Later, endogenous peroxidases were eliminated by the 10-min treatment with 3% H2O2, after which the slices were blocked in 5% bovine serum albumin (BSA; B928042, MACKLIN, China) for 30 min at 37°C. After that, the slices were incubated with a primary antibody against phosphorylated (p)-STAT1 (ab109461, Abcam, Cambridge, UK) at 4°C overnight, and with HRP-conjugated goat anti-rabbit IgG secondary antibody (31460, Thermo Fisher, USA) for 1 h in the dark. Areas positive for p-STAT1 were visualized by the addition of DAB solution (D8001, Sigma-Aldrich, USA). Counterstaining was conducted with hematoxylin (H9627, Sigma-Aldrich, USA). Staining results were observed via an optical microscope (CX31-LV320, Olympus, Tokyo, Japan) under 100 × magnification.
Co-immunoprecipitation (Co-IP) assay
Fresh mouse ileum from infected and control mice was homogenized and treated with T-PER histone protein extraction reagent (78510, Thermo Fisher, USA) to isolate protein lysate. Then the interaction between p-STAT1 and MUC4 was verified using Pierce Co-Immunoprecipitation kits (26149, Thermo Fisher, USA). In short, the isolated lysate was precleared by agarose resin, and then incubated overnight at 4°C with agarose resin coupled with normal Rabbit IgG (ab171870, Abcam, UK) or antibody for p-STAT1 (ab109461, Abcam, UK) or MUC4 (PA5-23077, Thermo Fisher, USA). Later, immunocomplexes were generated. After being eluted using elution buffer (21009, Thermo Fisher, USA), the immunocomplexes were subjected to Western blot, by which the enrichment of p-STAT1 or MUC4 was examined.
Western blot
Total protein was isolated from fresh mouse ileum using RIPA lysis buffer supplemented with protease and phosphatase inhibitors (PPC1010, Sigma-Aldrich, USA). After quantification using a BCA kit (A53227, Thermo Fisher, USA), the isolated proteins (30 µg) were denatured at 98°C for 10 min, and separated on 6% and 8% SDS-PAGE gel (P0686/P0688, Beyotime, China). The separated proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane (1620256, BIO-RAD, Hercules, CA, USA), and then blocked in 5% BSA for 1 h at room temperature. After washing with TBS-T (ab64204, Abcam, UK) three times, the membrane was incubated with primary antibodies for p-STAT1 (ab109461, 89 kDa, 1 : 1000, Abcam, UK), MUC4 (PA5-23077, 71 kDa, 1 : 5000, Thermo Fisher, USA) and GAPDH (ab8245, 37 kDa, 1 : 500, Abcam, UK) overnight at 4°C under gentle agitation. Later, the membrane was washed with TBS-T, followed by incubation with Goat anti-Rabbit/Mouse IgG secondary antibodies (ab97051/ab6728, Abcam, UK) for 2 h at room temperature. Protein bands were developed with Clarity Western ECL Substrate (1705060, BIO-RAD, USA) in an imaging system (LAS-3000, Fujifilm, Tokyo, Japan), and the band intensity was quantified using ImageJ software (3.0 version, National Institutes of Health, Bethesda, MA, USA).
Cell isolation, culture and transfection
Primary mouse intestinal epithelial cells (IECs) were isolated from healthy male C57BL/6 mice (n = 6) as previously described [39]. In brief, mouse small intestines were dissected and washed with a solution containing 0.154 M NaCl and 1mM DTT. Intestinal segments were incubated with phosphate buffered saline (PBS; P5493, Sigma-Aldrich, USA) at 37°C for 15 min. Then, the PBS used for incubation was supplemented with 1.5 mM EDTA (EDS, Sigma-Aldrich, USA) and 0.5 mM DTT, and incubated with the segments for an additional 30 min, followed by obtaining IECs.
The isolated IECs were grown in DMEM (11966025, Thermo Fisher, USA) supplemented with 10% fetal bovine serum (FBS; 12484028, Thermo Fisher, USA) and penicillin-streptomycin (15140122, 100 U/ml-100 µg/ml, Thermo Fisher, USA) on fibronectin-coated plates at 37°C with 5% CO2.
STAT1 overexpression plasmids were synthesized using pcDNA 3.1 (V79020, Thermo Fisher, USA), with empty vectors serving as the negative control (NC). pLKO.1-puro vectors (SHC016, Sigma-Aldrich, USA) were used to produce short hairpin RNA against MUC4 (shMUC4) (sense, 5′-UAUUAAUCUCUUAUCUUCCAC-3′; antisense, 5′-GGAAGAUAAGAGAUUAAUAGG-3′), and the negative control (shNC) of shMUC4 was set using empty pLKO.1-puro vectors. IECs were transfected with NC plus shNC or STAT1 overexpression plasmids plus shNC/shMUC4, with the aid of Lipofectamine 3000 transfection reagent (L3000015, Thermo Fisher, USA). Briefly, IECs were inoculated on 96-well plates at a density of 1 × 104 cells/well until the confluency of cells reached 80%. After dilution together with Opti-MEM and P3000 reagent, the plasmids and Lipofectamine 3000 transfection reagent were incubated together for 15 min at 37°C to generate gene-lipid complexes, which were later incubated with the cells for 48 h for transfection.
Cell infection
IECs seeded in 96-well plates were grown to produce an 80% confluent monolayer, and then challenged with 1 × 106 CFU/well EPEC for 1 h or 2.5 h (only for flow cytometry) at 37°C [40].
Cell Counting Kit (CCK)-8 assay
Transfected IECs were seeded into 96-well plates and challenged with EPEC. Then, after the cells were adherent to the plate, 10 µl of CCK-8 reagent (CA1210, Solarbio, Beijing, China) was added to each well, and incubated with cells for 2 h at 37°C. The OD of each well was read at 450 nm by a microplate reader (EMax Plus, Molecular Devices, Sunnyvale, CA, USA), and relative cell viability was calculated according to the formula: Cell viability (%) =(ODtreatment group-ODblank group)/(ODcontrol group-ODblank group) × 100.
Flow cytometry
Transfected IECs were subjected to apoptosis assay performed with Annexin V-FITC/PI Staining kits (ab14085, Abcam, UK). In brief, the cells were challenged with EPEC, and then digested for 10 min with 0.25% EDTA-free trypsin (T6325, MACKLIN, China). Centrifugation at 2000 × g was conducted for 10 min to obtain 1 × 105 cells. After being flushed with PBS, the cells were resuspended in Annexin V binding buffer, and then supplemented with Annexin V-FITC (5 µl) and propidium iodide (PI) (5 µl). Cell incubation was performed for 5 min protected from light at room temperature. The cells were later transferred onto a flow cytometer (Cytoflex, Beckman Coulter, Brea, CA, USA) for apoptosis detection, and data analysis was implemented using FlowJo software (Tree Star, Ashland, OR, USA).
Immunofluorescence assay
The deparaffinized mouse ileum slices from the immunohistochemical test were dehydrated and received antigen repair. Transfected IECs were infected, fixated in 4% paraformaldehyde for 15 min, and permeabilized using 0.3% Triton X-100 (X100, Sigma-Aldrich, USA) for 20 min. Both the slices and the cells were blocked in 5% BSA for 30 min at room temperature, and incubated at 4°C overnight with antibodies for MUC4 (PA5-23077, Rabbit, Thermo Fisher, USA) and ZO-1 (for tissues, 14-9776-82, Rat, Thermo Fisher, USA)/(for cells, 40-2200, Rabbit, Thermo Fisher, USA). Later, they were probed with secondary antibodies, Goat anti-Rabbit IgG (H+L) conjugated with Alexa Fluor 488 (A-11008, Thermo Fisher, USA) and Goat anti-Rat IgG (H+L) conjugated with Alexa Fluor 594 (A-11007, Thermo Fisher, USA), and then counterstained with DAPI (D9542, Sigma-Aldrich, USA). Images were developed using a confocal scanning microscope (ECLIPSE TI-SR, Nikon, Japan).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from fresh mouse ileum as well as IECs using Trizol reagent (15596026, Thermo Fisher, USA), and then the total RNA concentration was determined using a spectrophotometer (NanoDrop 2000, Thermo Fisher, USA). cDNA was generated using reverse transcription kits (K1622, Yaanda Biotechnology, Beijing, China). Next, cDNA was amplified on a PCR detection system (LightCycler 96, Roche, Indianapolis, IN, USA) with Eastep qPCR Master Mix (LS2062, Promega, Madison, WI, USA), with the primers listed in Table I, under the following thermocycle: 40 circles of 95°C for 10 min, 95°C for 15 s and 60°C for 1 min. Gene expression levels relative to the expression of the housekeeping gene GAPDH were calculated using the 2–ΔΔCt method [41].
Table I
Primers used in quantitative reverse transcription polymerase chain reaction for related genes
Statistical analysis
A total of 18 male C57BL/6 mice were included in these experiments. Statistical analysis was conducted on a sample size of 6 for EPEC-infected mice, 6 for control mice. The remaining 6 male C57BL/6 mice were used to isolate IECs, which were used for subsequent experiments, and three parallel experiments were set up for each group. GraphPad Prism (version 8.0, GraphPad Software Inc., San Diego, CA, USA) was used for statistical analysis. All data were presented as mean ± standard deviation (SD) from experiments repeated at least three times. Comparison between two groups was implemented via the independent-t test, while that among multiple groups was carried out using one-way analysis of variance (ANOVA). A difference was considered statistically significant when p < 0.05.
Results
p-STAT1, MUC4, ZO-1 and AMP-related genes showed low expression in the ileum of EPEC-infected mice
STAT1 downregulation has been reported to be concomitant with a reduction in AMP formation, resulting in intestinal microenvironmental disorders [22]. Moreover, AMPs are less distributed in mice bearing CRC as a result of MUC4 knockout [24]. We first investigated the expression of p-STAT1 and MUC4 in a mouse EPEC model. Immunohistochemical test results showed that in the ileum of EPEC-infected mice, p-STAT1 expression declined considerably (Figure 1 A). MUC4 was found to be lowly expressed in the ileum of EPEC-infected mice through immunofluorescence assay (Figure 1 B). We also found that this was accompanied by decreased expression of ZO-1 (Figure 1 C). Furthermore, Western blot analysis reaffirmed that the expression levels of p-STAT1 and MUC4 both diminished in the ileum of EPEC-infected mice (Figure 2 A, p < 0.001). Then, to determine how AMP level is affected in EPEC-induced intestinal infection, AMP-related gene expression levels were analyzed by qRT-PCR. The results revealed that the expression levels of Defa1, Defa4, Defa5, Camp and Lyz1 all diminished in the ileum of mice after EPEC infection (Figures 2 B–F, p < 0.001). In conclusion, we verified that the expression levels of p-STAT1, MUC4, ZO-1 and AMP-related gene were lower in the EPEC-infected mice, and STAT1 can positively regulate MUC4 expression [31]. Therefore, we next explored whether STAT1 and MUC4 could interact.
Figure 1
Phosphorylated-signal transducer and activator of transcription 1 (p-STAT1), mucin 4 (MUC4) and zonula occludens-1 (ZO-1) were lowly expressed in the ileum of enteropathogenic E. coli (EPEC)-infected mice. A–C – C57BL/6 mice were orally challenged with 100 µl of DMEM containing 1010 CFU/ml EPEC. A – Expression of p-STAT1 in the ileum was detected by immunohistochemical test (magnification: 100×; scale bar: 50 µm). B, C – Expression levels of MUC4 and ZO-1 in the ileum were determined by immunofluorescence assay (magnification: 400×; scale bar: 50 µm) (n = 6)
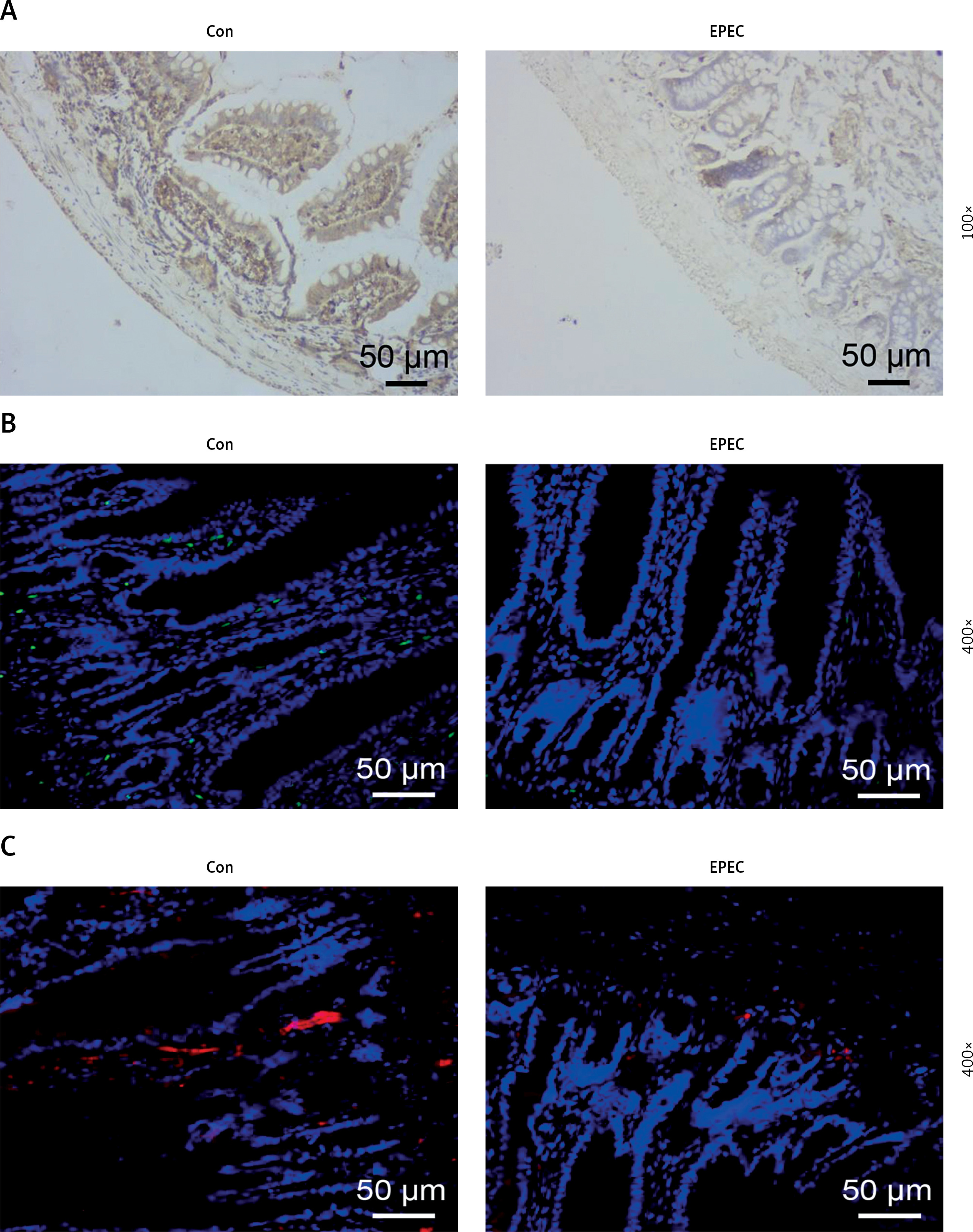
Figure 2
Antimicrobial peptide (AMP)-related gene expression levels decreased along with downregulation of the phosphorylated-signal transducer and activator of transcription 1 (p-STAT1)/mucin 4 (MUC4) complex in the ileum of enteropathogenic E. coli (EPEC)-infected mice. A–G – C57BL/6 mice were orally challenged with 100 µl of Dulbecco’s modified Eagle medium (DMEM) containing 1010 CFU/ml EPEC. A – Expression levels of p-STAT1 and MUC4 in the ileum were analyzed using Western blot, with GAPDH serving as the reference gene. B–F – Expression levels of Defa1, Defa4, Defa5, Camp and Lyz1 in the ileum were analyzed using quantitative reverse transcription polymerase chain reaction (qRT-PCR), with GAPDH serving as the reference gene. G – In the ileum, the binding of STAT1 to MUC4 was examined by co-immunoprecipitation assay. ###P < 0.001; #compared the values of the connected two groups (n = 6)
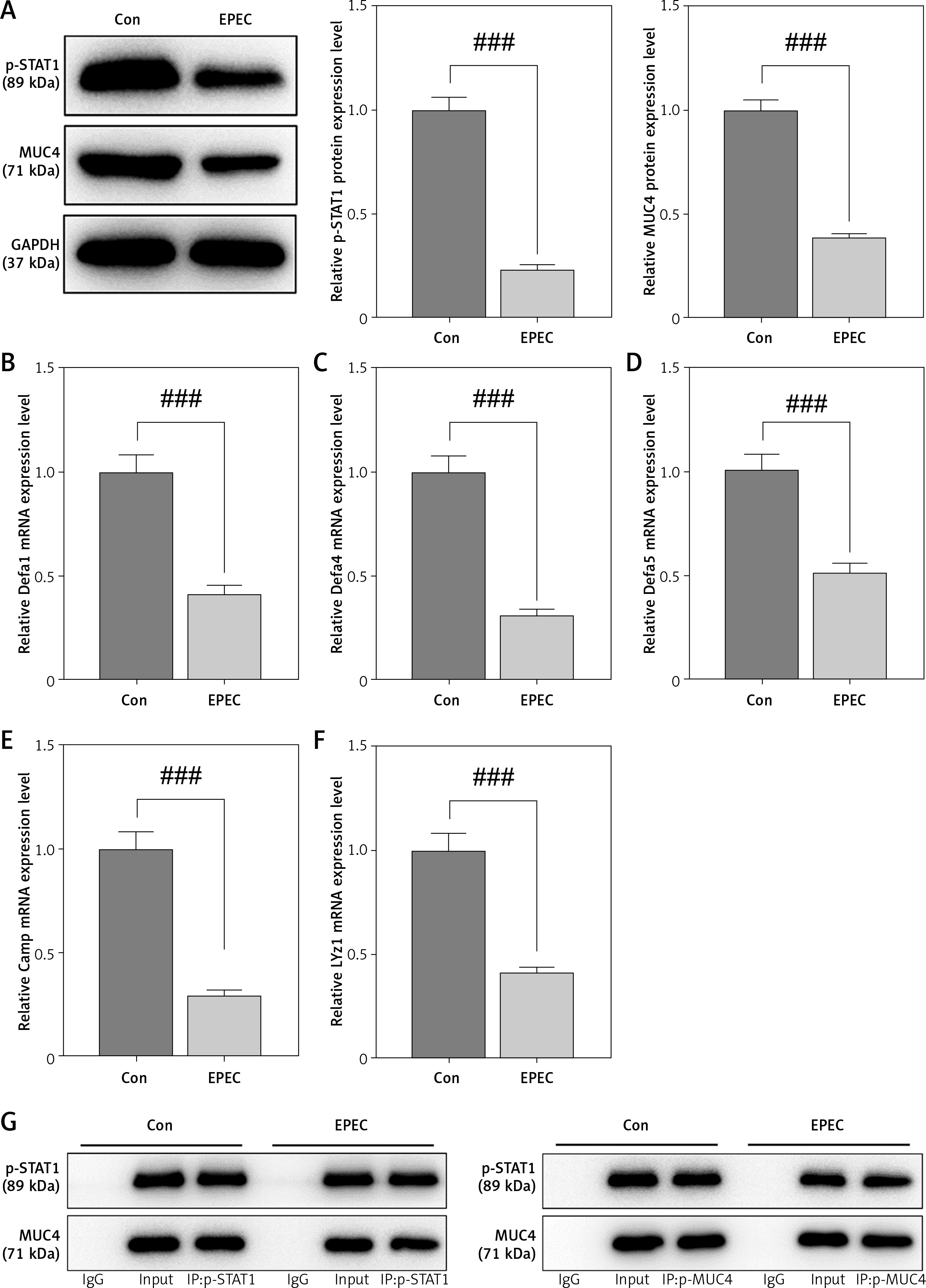
Co-IP assay was then performed to investigate the interaction between p-STAT1 and MUC4. The results proved that antibodies for p-STAT1 enriched MUC4 proteins and antibodies for MUC4 enriched p-STAT1 proteins in the ileum of both control and EPEC-infected mice (Figure 2 G). This indicated that p-STAT1 and MUC4 can interact with each other. Then, through in vitro experiments, we explored the STAT1/MUC4 pathway in relation to IEC viability, apoptosis and related inflammatory factors.
STAT1/MUC4 pathway was downregulated in EPEC-induced IECs and its forced upregulation resisted EPEC-induced IEC viability decreases
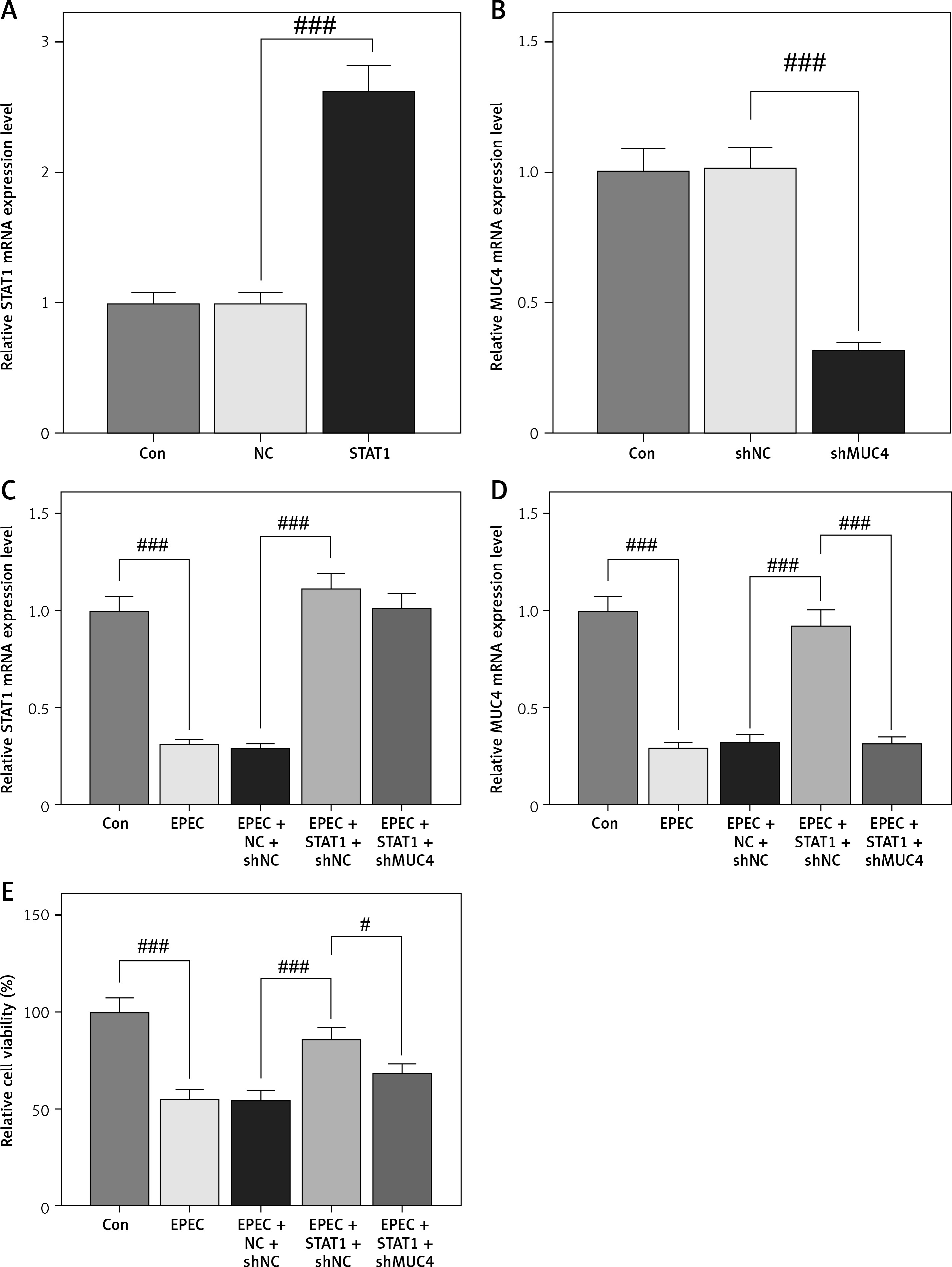
IECs were infected with EPEC to establish in-vitro intestinal infections, where the role of the STAT1/MUC4 pathway in EPEC-induced intestinal infections was examined. Transfection with STAT1 overexpression plasmids led to increased expression of STAT1 (Figure 3 A, p < 0.001), and MUC4 expression was knocked down by transfection with shMUC4 (Figure 3 B, p < 0.001) in IECs. STAT1 expression declined with EPEC infection in IEC (Figure 3 C, p < 0.001), which was reversed by STAT1 overexpression plasmid transfection (Figure 3 C, p < 0.001). MUC4 knockdown had no significant impact on the transfection-mediated STAT1 overexpression in EPEC-infected IECs (Figure 3 C). EPEC infection resulted in MUC4 downregulation in IECs (Figure 3 D, p < 0.001). STAT1 overexpression abrogated EPEC infection-induced MUC4 downregulation (Figure 3 D, p < 0.001), while such an effect of STAT1 overexpression was attenuated by MUC4 knockdown (Figure 3 D, p < 0.001). Moreover, decreased viability of IECs after EPEC infection was detected (Figure 3 E, p < 0.001). STAT1 overexpression resisted the EPEC-induced IEC cell viability decrease (Figure 3 E, p < 0.001); nevertheless, MUC4 knockdown mitigated the enhancing effect of STAT1 overexpression on the viability of EPEC-infected IECs (Figure 3 E, p < 0.05).
Figure 3
Signal transducer and activator of transcription 1 (STAT1)/mucin 4 (MUC4) pathway was downregulated in enteropathogenic E. coli (EPEC)-induced mouse intestinal epithelial cells (IEC) and its forced upregulation resisted EPEC-induced IEC viability decrease. A, B – The expression levels of STAT1 and MUC4 in IECs transfected with STAT1 overexpression plasmids/negative control (NC) or short hairpin RNA against MUC4 (shMUC4)/short hairpin RNA against negative control (shNC) were analyzed using quantitative reverse transcription polymerase chain reaction (qRT-PCR), with GAPDH serving as the reference gene. C–E – IECs were transfected with NC plus shNC or STAT1 overexpression plasmids plus shNC/shMUC4, followed by EPEC (1 × 106 CFU/well) infection. C, D – Expression levels of STAT1 and MUC4 in the IECs were analyzed using qRT-PCR, with GAPDH serving as the reference gene. E – The viability of the IECs was measured by Cell Counting Kit-8 assay. #p < 0.05; ###p < 0.001; #compared the values of the connected two groups (n = 3)
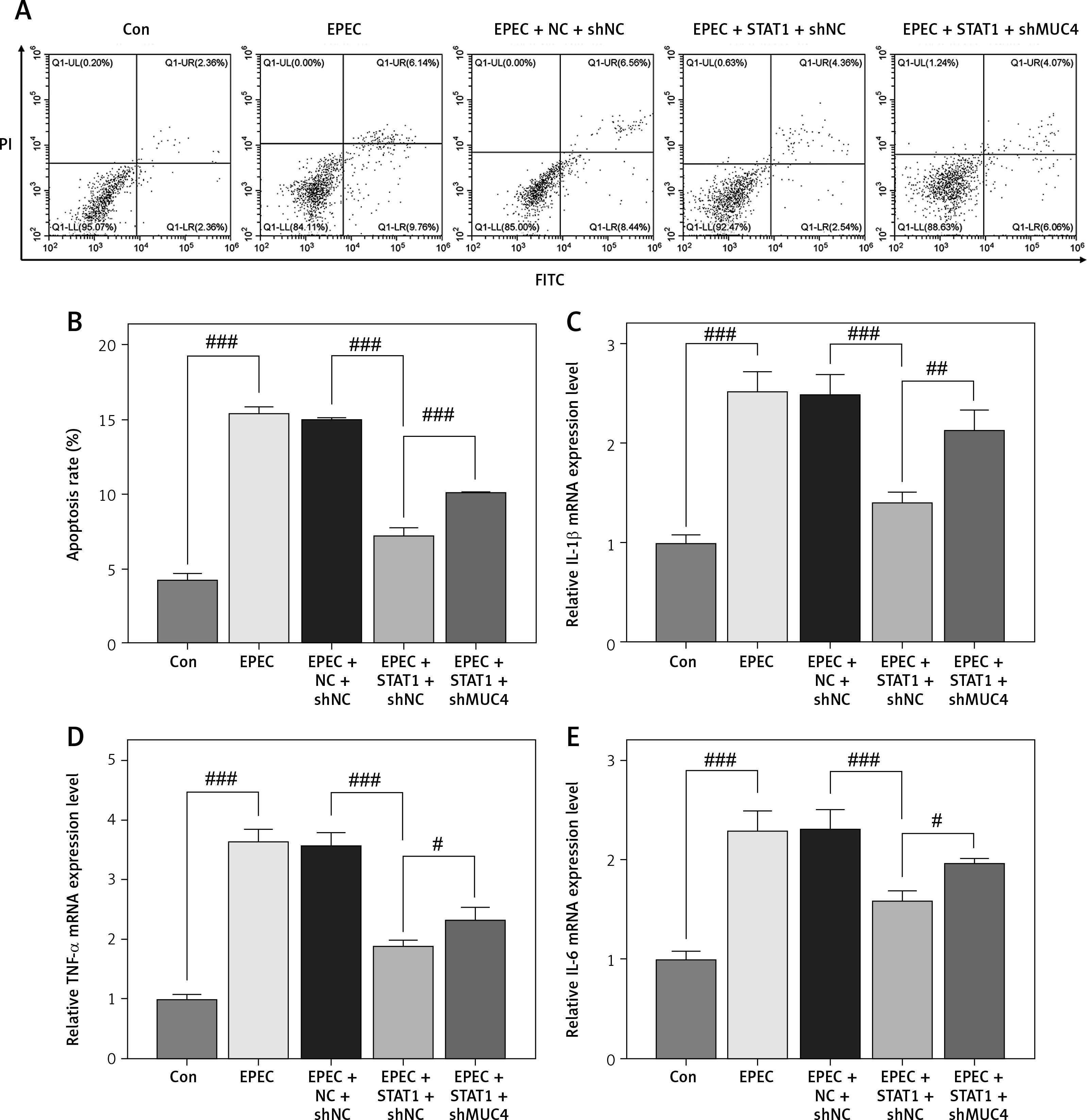
Forced upregulation of STAT1/MUC4 pathway offset EPEC-induced apoptosis promotion and release of proinflammatory cytokines in IEC
Subsequently, EPEC infection was observed to induce apoptosis of IECs (Figures 4 A, B, p < 0.001), which was offset by STAT1 overexpression (Figures 4 A, B, p < 0.001). MUC4 knockdown attenuated the inhibiting effect of STAT1 overexpression on apoptosis of EPEC-infected IECs (Figures 4 A, B, p < 0.001). In addition, the levels of proinflammatory cytokines (IL-1, TNF-α and IL-6) rose in IEC following EPEC infection (Figures 4 C–E, p < 0.001). STAT1 overexpression caused downregulation of IL-1, TNF-α and IL-6 in EPEC-infected IECs (Figures 4 C–E, p < 0.001), but the downregulation was mitigated by MUC4 knockdown (Figures 4 C–E, p < 0.05).
Figure 4
Forced upregulation of signal transducer and activator of transcription 1 (STAT1)/mucin 4 (MUC4) pathway offset enteropathogenic E. coli (EPEC)-induced apoptosis promotion and release of proinflammatory cytokines in IEC. A–E – mouse intestinal epithelial cells (IECs) were transfected with negative control (NC) plus short hairpin RNA against negative control (shNC) or STAT1 overexpression plasmids plus shNC/short hairpin RNA against MUC4 (shMUC4), followed by EPEC (1 × 106 CFU/well) infection. A, B – Apoptosis of the IECs was detected by flow cytometry. C–E – Expression levels of interleukin-1β (IL-1 β), tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) in the IECs were analyzed using quantitative reverse transcription polymerase chain reaction (qRT-PCR), with GAPDH serving as the reference gene. #p < 0.05; ##p < 0.001; ###p < 0.001; # compared the values of the connected two groups (n = 3)
PI – propidium iodide, FITC – fluorescein isothiocyanate
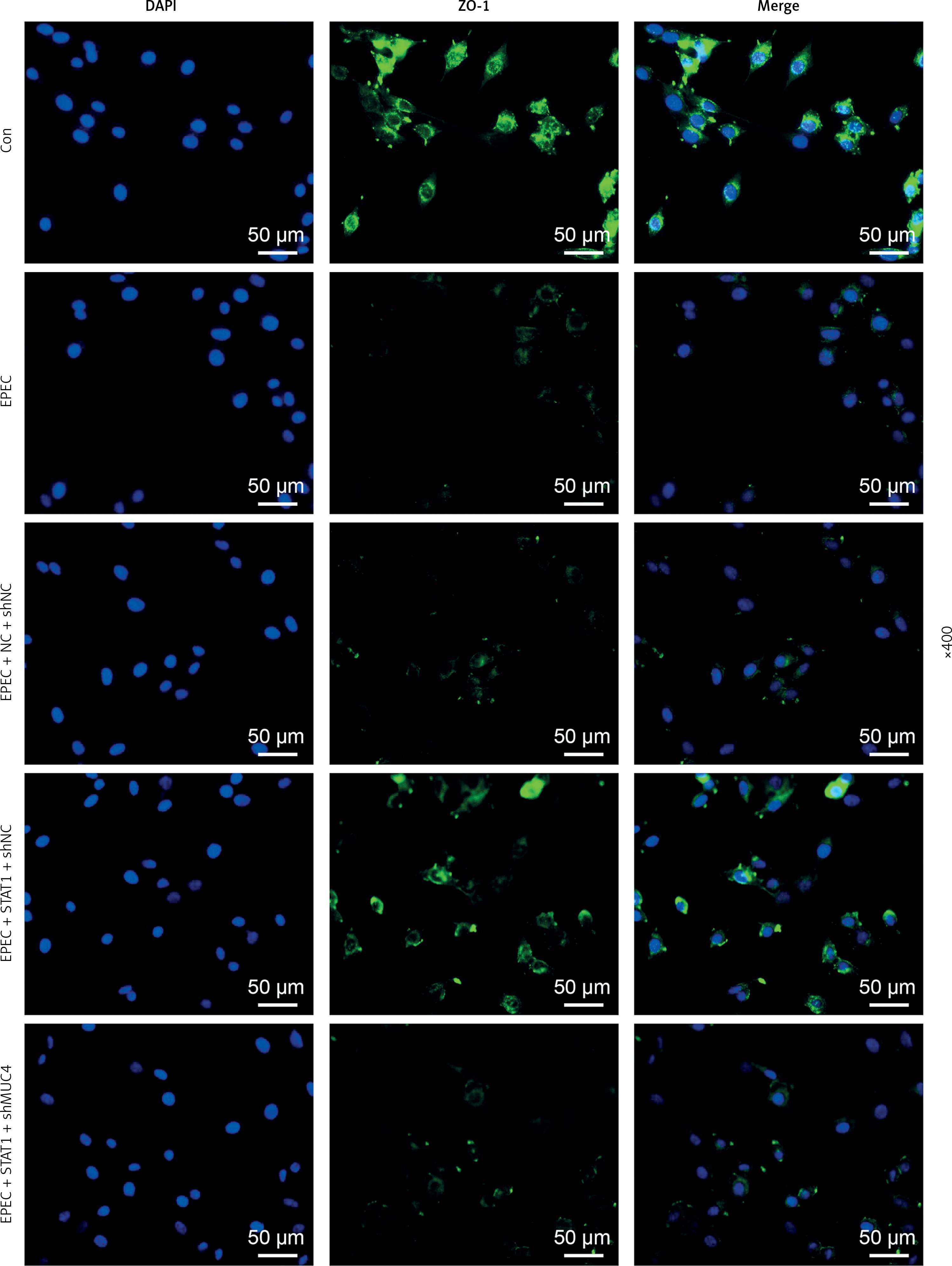
Forced upregulation of STAT1/MUC4 pathway resisted EPEC-induced ZO-1 downregulation in IEC
Through immunofluorescence assay, the expression of ZO-1 was detected to be reduced by EPEC infection in IEC, and this ZO-1 expression reduction could be abrogated by STAT1 overexpression (Figure 5). MUC4 knockdown weakened the effect of STAT1 overexpression on ZO-1 expression in EPEC-infected IECs through suppressing ZO-1-positive fluorescence (Figure 5).
Figure 5
Forced upregulation of signal transducer and activator of transcription 1 (STAT1)/mucin 4 (MUC4) pathway resisted enteropathogenic E. coli (EPEC)-induced zonula occludens-1 (ZO-1) downregulation in mouse intestinal epithelial cells (IEC). IECs were transfected with negative control (NC) plus short hairpin RNA against negative control (shNC) or STAT1 overexpression plasmids plus shNC/short hairpin RNA against MUC4 (shMUC4), followed by EPEC (1 × 106 CFU/well) infection, and the expression of ZO-1 in the IECs was determined by immunofluorescence assay (magnification: × 400; scale bar: 50 µm) (n = 3)
DAPI – 4’,6-diamidino-2-phenylindole.
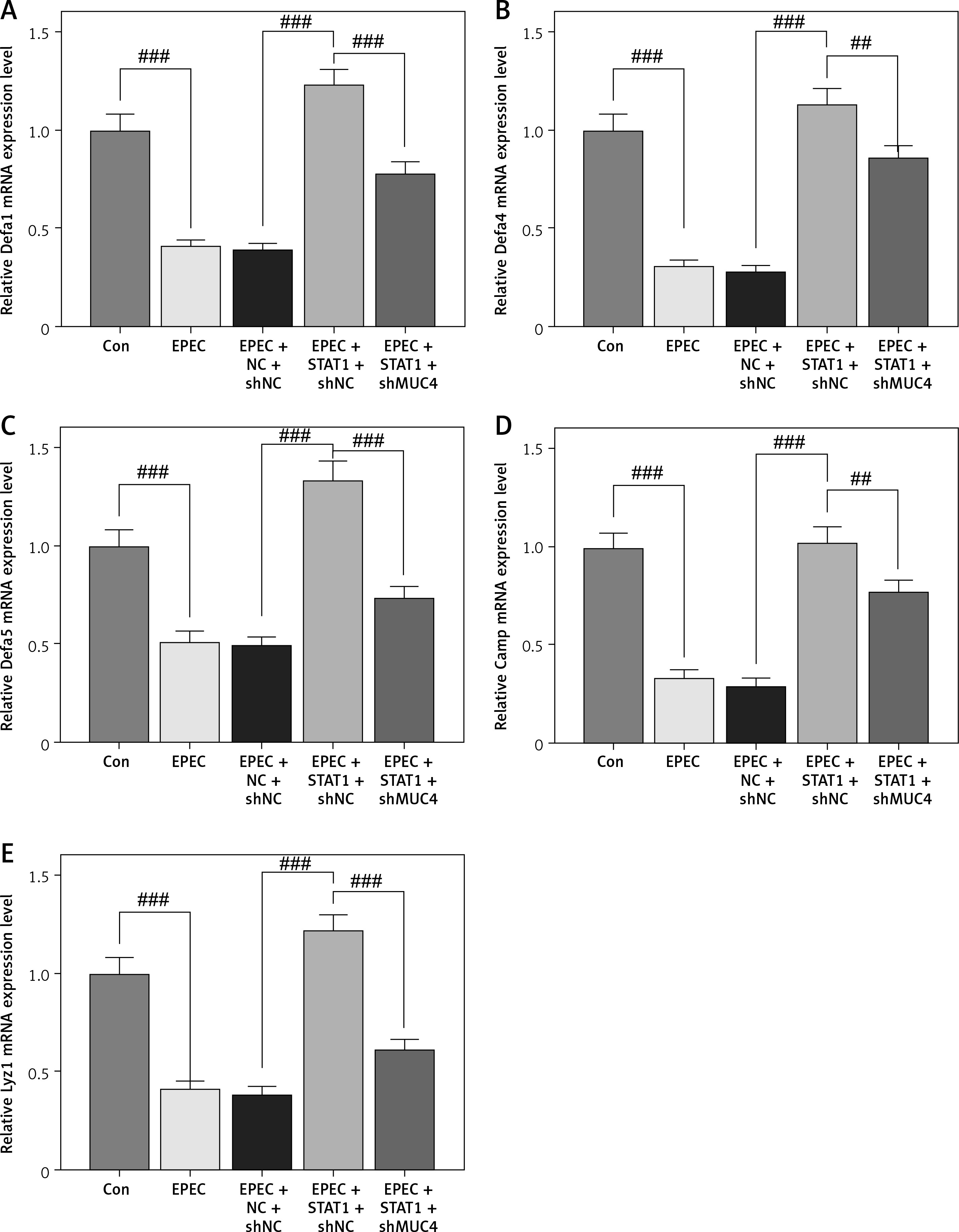
Forced upregulation of STAT1/MUC4 pathway reversed EPEC-induced AMP-related gene downregulation in IEC
Finally, whether and how the STAT1/MUC4 pathway influences AMP formation in in-vitro intestinal infection were investigated. Upon EPEC infection, the expression levels of AMP-related genes (Defa1, Defa4, Defa5, Camp and Lyz1) all decreased in IECs (Figures 6 A–E, p < 0.001). STAT1 overexpression observably upregulated the expression levels of these AMP-related genes, and reversed the EPEC-induced negative effect on AMP formation in IECs (Figures 6 A–E, p < 0.001). However, MUC4 knockdown offset the STAT1 overexpression-induced upregulation of these AMP-related genes in EPEC-infected IECs (Figures 6 A–E, p < 0.01).
Figure 6
Forced upregulation of signal transducer and activator of transcription 1 (STAT1)/mucin 4 (MUC4) pathway reversed enteropathogenic E. coli (EPEC)-induced antimicrobial peptide (AMP)-related gene downregulation in mouse intestinal epithelial cells (IEC). A–E – IECs were transfected with negative control (NC) plus short hairpin RNA against negative control (shNC) or STAT1 overexpression plasmids plus shNC/short hairpin RNA against MUC4 (shMUC4), followed by EPEC (1 × 106 CFU/well) infection, and the expression levels of Defa1, Defa4, Defa5, Camp and Lyz1 in the ileum were analyzed using quantitative reverse transcription polymerase chain reaction (qRT-PCR), with GAPDH serving as the reference gene. ##p < 0.001; ###p < 0.001; #compared the values of the connected two groups (n = 3)
Discussion
EPEC infection remains a public health concern in developing countries, leading to diarrheal diseases with a risk of death [42]. AMPs can act as endogenous antibiotics to direct bactericidal activity, thereby reducing epithelial barrier injury caused by EPEC infection [43]. In the present study, the STAT1/MUC4 pathway was identified as the upstream mechanism of AMP-induced protection against EPEC infection in intestinal epithelial barriers.
Inhibited STAT1 signaling is associated with the exacerbation of enteropathogenic bacterial infection-caused intestinal hyperplasia and diarrhea in mice [44]. In mouse models of chronic alcohol-induced intestinal injury, lack of p-STAT1 and of total STAT1 in the ileum cooccurs with downregulation of small intestinal AMPs, resulting in disrupted gut microbiota homeostasis [22]. The above findings suggested that STAT1 downregulation or inactivation may be associated with microbiota homeostasis disruption following bacterial infection. Consistently, our study showed that STAT1 phosphorylation diminished in the ileum of mice following EPEC infection. In contrast, Ceponis et al. found that EPEC infection in epithelial cells could not eliminate STAT1 tyrosine phosphorylation induced by IFN-γ [45]. However, EPEC exhibits considerable genomic diversity, being represented in at least 10 different phylogenomic lineages [46], so EPEC with different phenotypes may have different effects. In our study, the STAT1 mRNA level declined in IEC after EPEC infection, which, together with Ceponis’s results, implies that EPEC infection reduces STAT1 mRNA expression but is unable to affect STAT1 phosphorylation. Therefore, the decrease of STAT1 phosphorylation in the ileum of mice following EPEC infection may be attributed to the decreased level of STAT1 designated to undergo phosphorylation. This also justifies the discrepancy between our and Ceponis’s results. Furthermore, IFN-γ, which can elicit STAT1 activation, is an important cytokine against infection by viral and microbial pathogens in host defense [47], and is induced in mice and humans following EPEC injection [48, 49]. The EPEC burden is higher in immunocompromised patients than in healthy adults [50]. These observations suggested that in healthy adults, host defense is activated to upregulate IFN-γ, which activates STAT1 and thus prevents EPEC infection.
The attachment of EPEC on the epithelium disrupts the normal cellular process [51]. EPEC encodes a subset of effectors that induce apoptosis of epithelial cells to cause cell death and barrier dysfunction [52, 53]. Moreover, inflammation of the intestinal mucosa occurs after EPEC infection, where EPEC is involved in stimulation of the proinflammatory cytokine TNF-α production to promote myosin light chain (MLC) phosphorylation, thereby contributing to barrier dysfunction [8, 54]. Also, following EPEC infection, rabbit enterocytes and rat colonic mucosa scrapings have been discovered to exhibit upregulated proinflammatory cytokines, IL-1β and IL-6 [55, 56], whose release is mediated by TNF-α [57]. In our study, we overexpressed STAT1 in order to achieve prominent STAT1 activation in IECs, and found that EPEC-induced apoptosis and upregulation of TNF-α, IL-1β and IL-6 were weakened by STAT1 overexpression.
Epithelial barrier dysfunction manifests alteration of tight junctions [43], which act as the barrier required to maintain polarity that aids the directional diffusion of solutes [58]. ZO-1 is a cytoplasmic scaffolding protein that constitutes tight junctions along with other membrane proteins [59], and plays a dispensable role for barrier function [60]. In our study, low ZO-1 expression caused by EPEC infection was reversed by STAT1 overexpression in IECs. The above results collectively indicated that STAT1 overexpression mitigated EPEC-induced intestinal epithelial barrier dysfunction.
In addition, Paneth cells are specialized cells in the intestinal epithelium [14], which assist the intestinal mucosa to maintain the functional intestinal epithelial barrier by secreting AMPs to modulate innate immunity [61], or directly kill their target bacteria [62]. Our study showed that in the ileum of mice and IECs following EPEC infection, p-STAT1 and STAT1 downregulation was concomitant with reduction of AMP formation, as reflected by decreased levels of AMP-related genes (Defa1, Defa4, Defa5, Camp and Lyz1), which is in line with their status in alcohol-induced intestinal injury [22]. Moreover, we observed that STAT1 overexpression facilitated AMP formation. AMPs are considered as therapeutic agents for treating infections caused by untreatable microorganisms. According to this and our results above, STAT1 activation is presumed to prevent EPEC infection by facilitating AMP formation.
The expression of MUC4, a member of the transmembrane mucin family, increases along with decreased epithelial paracellular permeability [63]. STAT1 can transcriptionally upregulate MUC4 level [31], which was reaffirmed in our study. Also, MUC4 protects epithelial surfaces against infections and injury [64]. MUC4 is lowly expressed in intestinal epithelial cells from patients with inflammatory bowel disease [65]. Notably, knockout of MUC4 leads to less AMP formation in CRC mice [24]. Our study revealed that MUC4 level decreased with STAT1 phosphorylation inhibition in the ileum of mice and IECs after EPEC infection, and MUC4 knockdown offset the effects of STAT1 overexpression on apoptosis, inflammation, and AMP formation in EPEC-infected IEC.
Previous research also demonstrated that JAK/STAT-1 plays a critical role in the regulation of uropathogenic E. coli invasion [66, 67]. Additionally, the oral antimicrobial peptide Mastoparan X alleviates intestinal inflammation caused by enterohemorrhagic E. coli and regulates the gut microbiota [68]. Moreover, E. coli is an important cause of spontaneous bacterial peritonitis (SBP), which is one of the major complications of liver cirrhosis, whose clinical symptoms include diarrhea. Enhancing intestinal barrier function is a novel strategy for the treatment of cirrhosis, as impaired intestinal barrier function can lead to translocation of intestinal bacteria [69]. Therefore, further studies are required to determine whether the mechanism of STAT1/MUC4 activation promotes antimicrobial peptide production to reduce intestinal epithelium barrier injury in other types of diarrhea-induced microorganisms. Furthermore, noncommunicable diarrhea can be caused by toxins, chronic diseases, or antibiotics [70]. Whether this mechanism also exists in non-infectious diarrhea needs further study.
There are some limitations in this study. EPEC may change the intestinal flora, and we did not study the effects of STAT1/MUC4 on the microbiota, which should be studied in the future. In addition, whether STAT1/MUC4 affects intestinal epithelium barrier injury and will cause translocation of microorganisms needs further research.
In conclusion, the present study demonstrated that the STAT1/MUC4 pathway is downregulated in intestinal epithelium after EPEC infection, and its activation facilitates AMP formation, partaking in the prevention of EPEC-induced intestinal epithelium infection.