The supraphysiological levels of cortisol seen in both ACTH-dependent and ACTH-independent Cushing’s syndrome lead not only to adverse metabolic consequences – such as hyperglycemia and weight gain – but also to profound immunosuppressive effects [1]. The degree of immunosuppression correlates directly with both the duration and severity of excess cortisol and, in severe cases, may predispose individuals to opportunistic infections. These well-documented immunosuppressive properties of glucocorticoids – including the suppression of abnormal immune responses and the promotion of immune tolerance – are widely used in clinical practice to treat a range of autoimmune and inflammatory diseases. Successful treatment of endogenous hypercortisolism usually results in a rapid decrease in circulating cortisol levels. Until the hypothalamic–pituitary–adrenal (HPA) axis returns to normal, patients require glucocorticoid replacement therapy, most commonly with hydrocortisone. However, physiologic replacement doses are not immunosuppressive, which may allow latent autoimmune processes to be unmasked or reactivated [2].
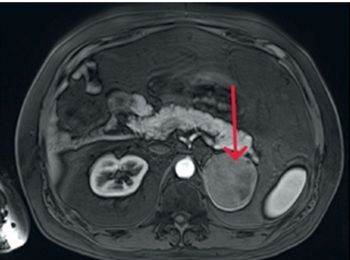
We present the case of a 42-year-old man admitted to the Department of Endocrinology and Metabolic Diseases with a 6-month history of weight gain, hypertension, lower back pain, and muscle weakness. He had already been diagnosed with a left adrenal mass, which was accidentally discovered during magnetic resonance imaging (MRI) of the lumbar spine. Physical examination revealed features of Cushing’s syndrome: moon face, facial plethora, supraclavicular fat pads, central obesity and abdominal purple striae. The aldosterone/renin ratio and urinary fractionated metanephrine measurements were within normal ranges. We observed elevated midnight cortisol (13.4 mg/dl) and UFC (432.9 mg/24 h). The lack of cortisol suppression in the high-dose DXM test alongside low plasma ACTH indicated ACTH-independent Cushing’s syndrome. Dehydroepiandrosterone level was within the normal range but relatively high. Computed tomography of the thorax, abdomen, and pelvis showed a left adrenal mass, which was 75 × 68 mm in size (Figure 1). Its radiological features did not suggest a benign lesion, and there were no visible metastases or lymphadenopathy, which was consistent with the MRI findings (Figures 2, 3). Given the clinical picture, adrenocortical carcinoma was suspected; therefore, open adrenalectomy was performed. The postoperative period was uneventful, and the patient was discharged from hospital with the recommendation for corticosteroid replacement therapy. The pathology report revealed the mass to be an adrenocortical neoplasm of uncertain malignant potential (modified Weiss score of 2, Weiss score of 3, Helsinki score of 2 and Ki-67 proliferation index < 1). Close postoperative follow-up was implemented, including biochemical evaluation and computed tomography (CT) imaging. At the first follow-up, his cushingoid features resolved, with a decreasing serum cortisol concentration. The corticotropin stimulation test indicated adrenocortical insufficiency. CT imaging was negative for adrenal tumor recurrence; however, CT scan of the chest showed hilar and mediastinal lymphadenopathy (Figure 4). The patient was referred to the pulmonology department and underwent EBUS. Histopathology was consistent with sarcoidosis, with no evidence of malignancy. There was no indication for sarcoidosis-specific treatment, and the patient remained on hydrocortisone replacement therapy. At the second follow-up visit 5 months later, the patient reported overall well-being, although he complained of persistent muscle weakness and mild arthralgia – symptoms that may reflect cortisol deficiency, steroid withdrawal syndrome, or ongoing sarcoidosis activity. He reported no fever, malaise, visual disturbances, or dyspnea. Laboratory tests showed elevated angiotensin-converting enzyme levels and mild PTH-independent hypercalcemia. Repeat CT scans showed no significant changes compared to previous assessments. As a result, vitamin D supplementation was discontinued, and the hydrocortisone dosage was maintained. During 13 months of follow-up, there was no evidence of recurrence of Cushing’s syndrome, and complete remission of sarcoidosis was confirmed. This case illustrates a phenomenon increasingly noted in the literature: the emergence or unmasking of immune-mediated diseases after successful treatment of endogenous hypercortisolemia. Glucocorticoids affect both innate and adaptive immunity by modulating gene expression, cytokine signaling, and immune cell activity. Chronic hypercortisolemia suppresses these immune pathways, and their reactivation after treatment can trigger autoimmune manifestations [3].
Figure 1
Computed tomography image, axial projection, native image; blue arrow indicates a focal lesion of the left adrenal gland
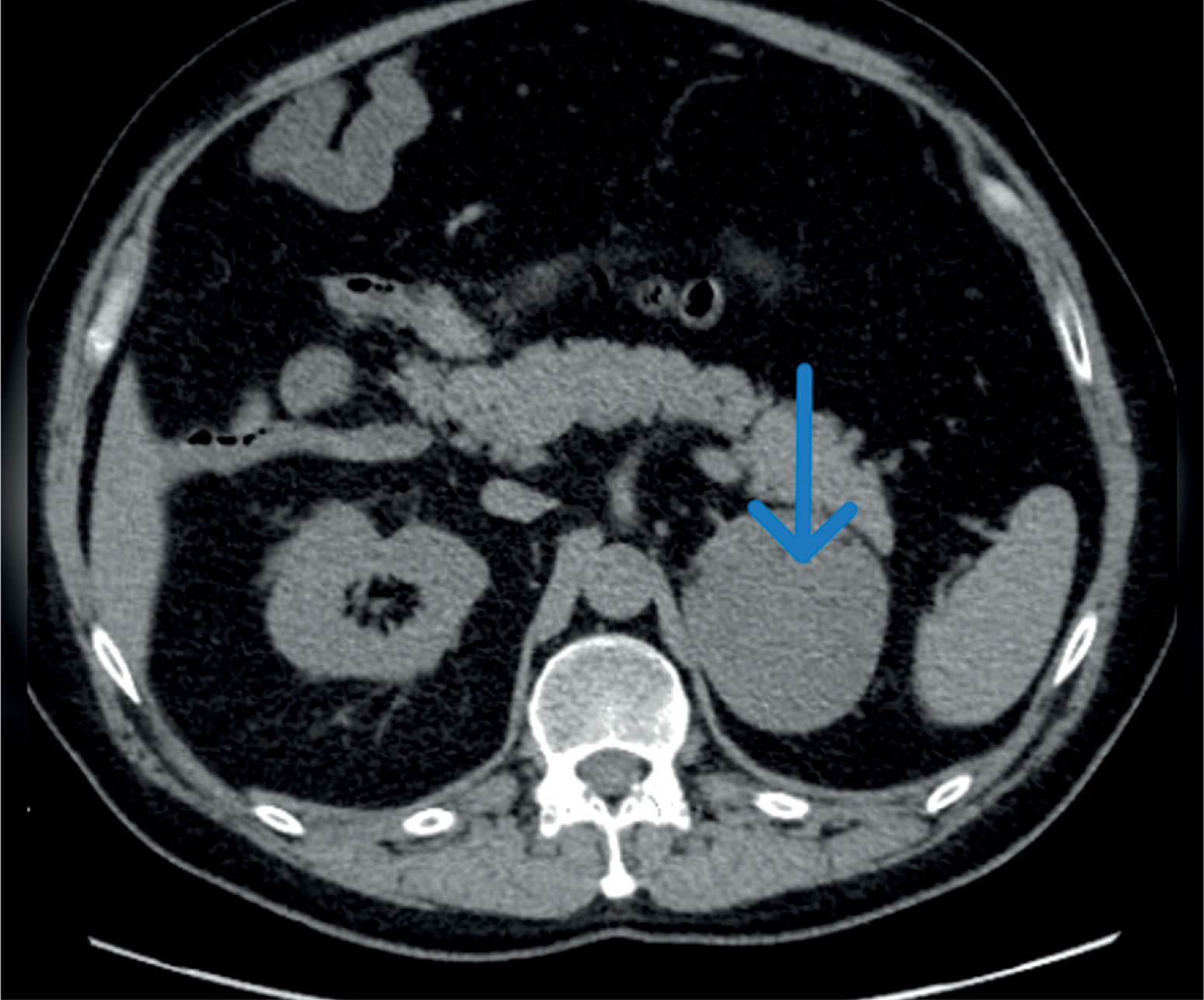
Figure 2
The magnetic resonance imaging demonstrates a large tumor between the spleen and right kidney; the kidney is displaced caudally
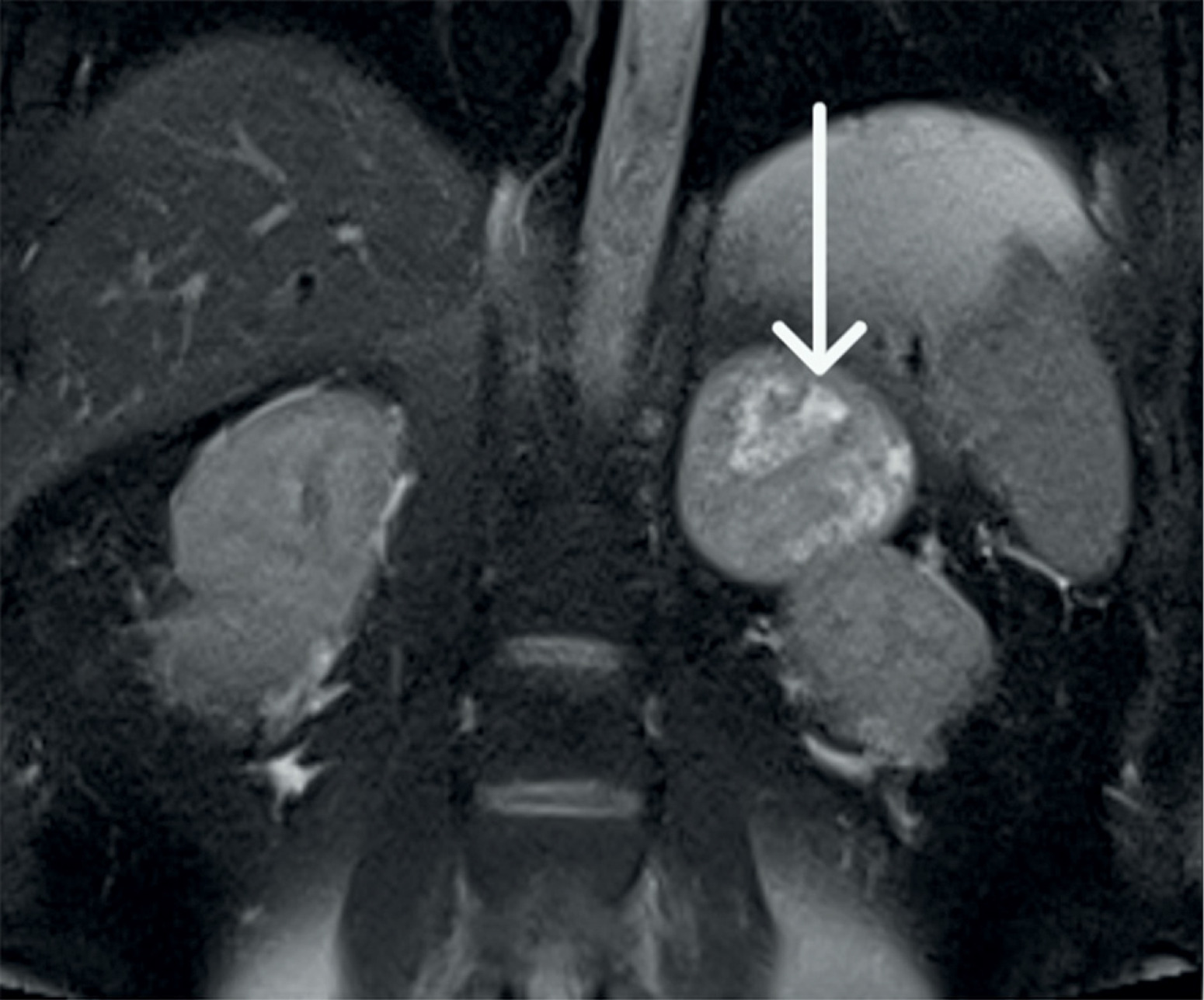
Figure 3
The magnetic resonance imaging demonstrates a large tumor between the right kidney and the pancreas. Following contrast, the lesion demonstrates heterogeneous peripheral enhancement
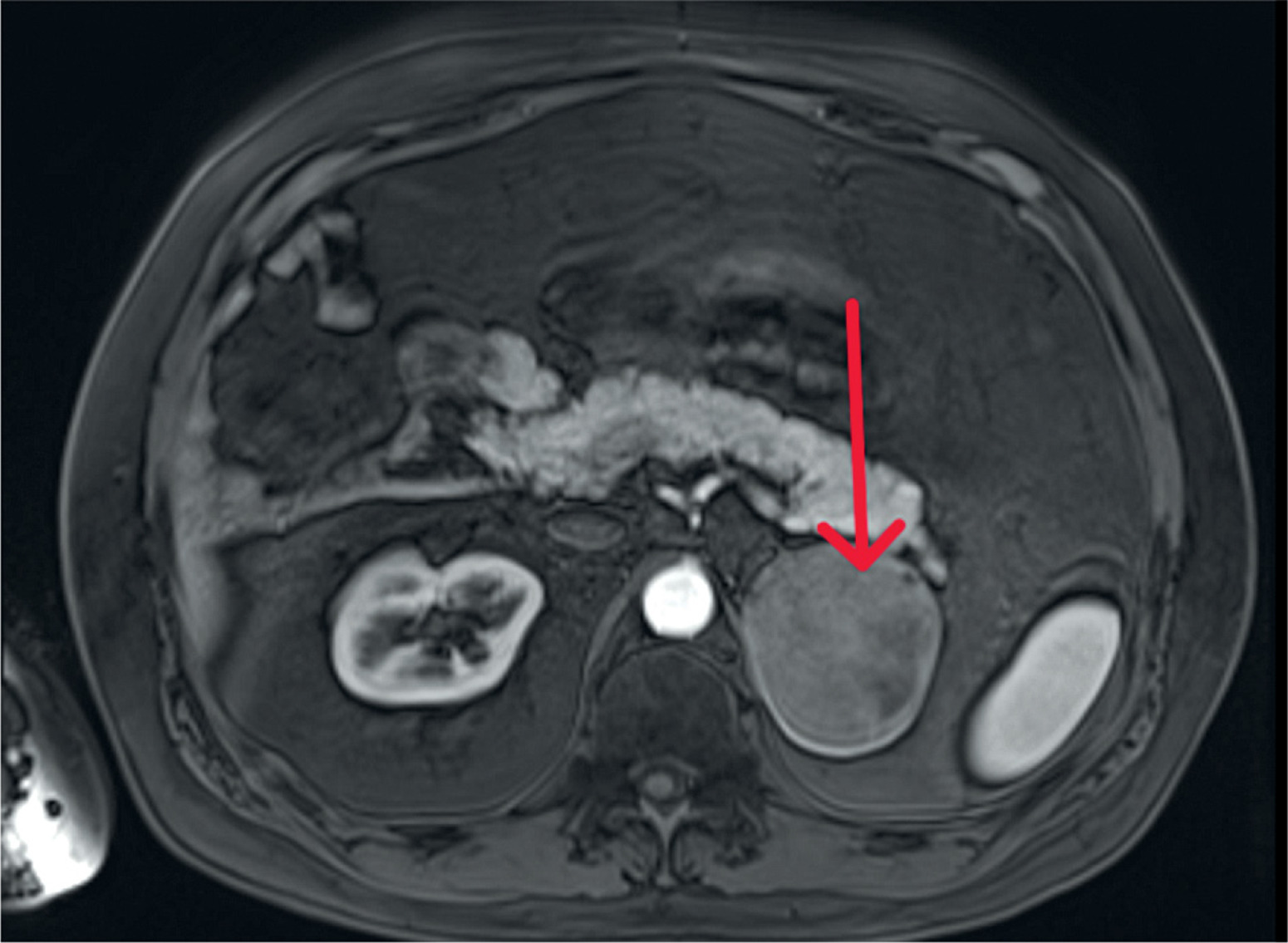
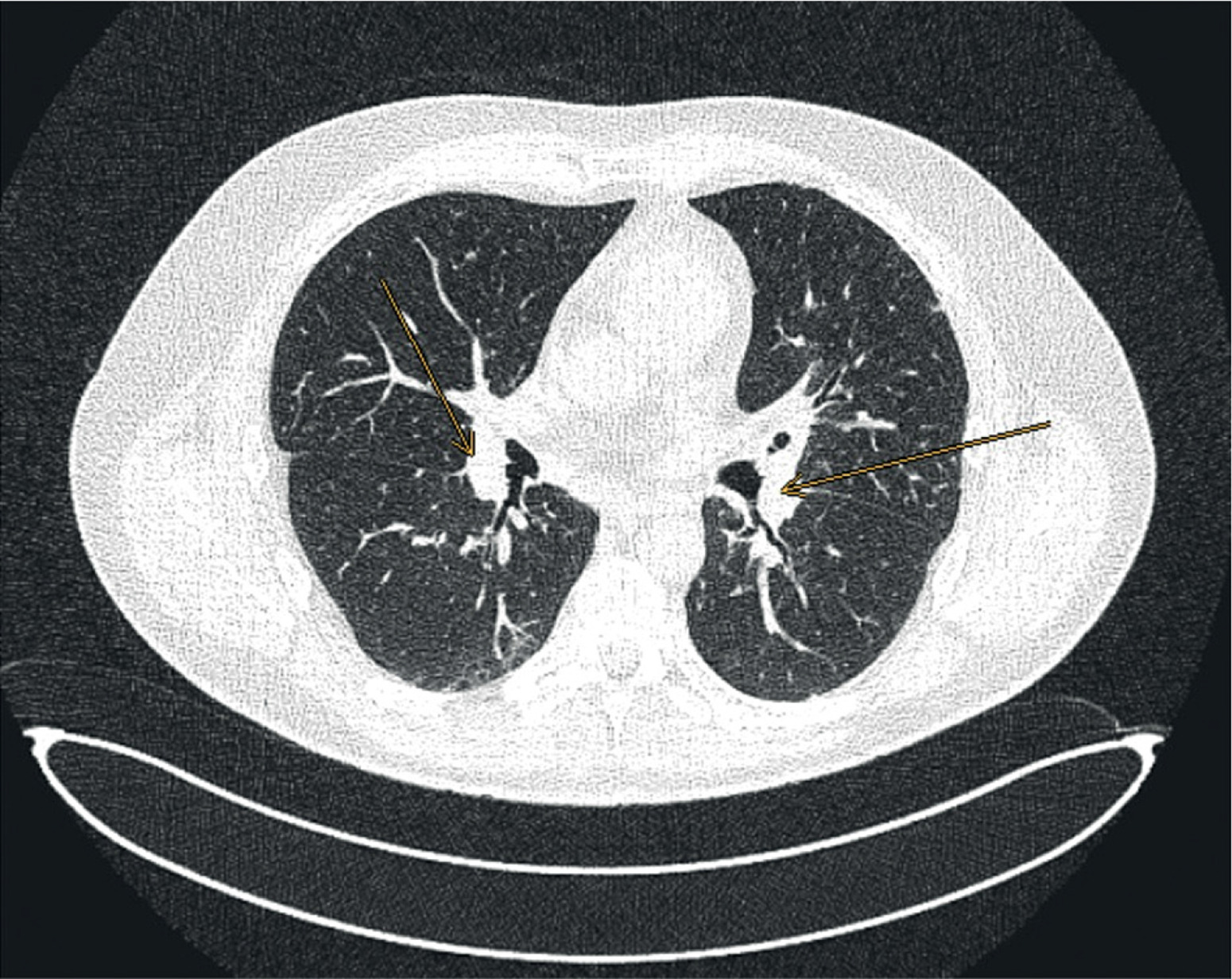
Figure 4
Computed tomography image, axial projection, lung window; orange arrows indicate enlarged hilar lymph nodes
Literature reports describe a wide spectrum of immunological disorders that appear after hypercortisolemia has subsided. The most frequently observed de novo conditions after remission, regardless of etiology, include thyroid disorders such as autoimmune thyroiditis and Graves’ disease. In addition, several cases of rheumatologic diseases have been documented, including systemic lupus erythematosus, rheumatoid arthritis, seronegative arthritis, polymyalgia rheumatica, giant cell arteritis, and retinal vasculitis. Isolated case reports also describe neurological conditions with presumed autoimmune etiology, such as multiple sclerosis, lymphocytic hypophysitis, and myasthenia gravis [1]. In addition, several reports have highlighted the emergence of novel dermatologic manifestations, such as psoriasis and angioedema, as well as immune-mediated gastrointestinal disorders, including celiac disease and autoimmune hepatitis [2]. Our report adds another case of sarcoidosis to the growing literature on immune-mediated diseases that occur after hypercortisolism has resolved. It highlights the potential role of immune dysregulation in the pathogenesis of sarcoidosis, a granulomatous disease of unknown etiology that often involves intrathoracic lymph nodes and lung tissue. The diagnosis of sarcoidosis in our patient shortly after adrenalectomy, coinciding with the abrupt cortisol withdrawal, strongly supports the immune rebound hypothesis [4–6]. This case highlights the importance of increased clinical vigilance during the post-remission period of hypercortisolemia. The presenting symptoms may mimic adrenal insufficiency or glucocorticoid withdrawal syndrome, complicating the diagnostic evaluation. Clinicians should be aware of this potential overlap and consider immune-mediated conditions in the differential diagnosis when evaluating patients with nonspecific systemic complaints after resolution of Cushing’s syndrome.