Introduction
Androgen insensitivity syndrome (AIS) is an X-linked recessive genetic condition, characterized by malformations that appear as a result of an androgen receptor (AR) mutation. This mutation interferes with and disrupts the normal interaction between androgens and their receptors. Androgen insensitivity syndrome implies the existence of a partial or total lack of response or sensitivity of androgen receptors to androgen stimulation [1]. This alters the masculinization process in chromosomal male patients (46XY karyotype) and will lead to the development of a phenotype that can range from normal female external genitalia to normal male external genitalia, but with various degrees of infertility and gynecomastia. These anatomical characteristics are consistent with the degree of androgen receptor sensitivity to the androgen stimulation [2]. Complete, partial, and mild androgen insensitivity syndromes fall into the generic category of 46XY disorder of sex development.
In the literature data, the first descriptions of the effects of AIS took the form of comprehensive descriptions of intersex characteristics or individual case reports [3, 4]. Since the 16th century, descriptions, criteria and taxonomies for the classification of intersexuality have been developed by physicians of the time [3–6]. Scientists have used the term “hermaphrodite” as the basis of their taxonomies. The first full description of the AIS dates back to the mid-20th century, when Morris presented 82 female patients with bilateral testes; he initially described this condition as “testicular feminization syndrome” [7]. Later, as a consequence of a better understanding of the pathophysiology involved, the syndrome was renamed AIS [1], which is the currently accepted medical terminology.
Androgen insensitivity syndrome has complex and varied clinical manifestations which pose many challenges for physicians and patients. The clinical manifestations may be specific to each phenotype, depending on the degree of androgen insensitivity.
Material and methods
This narrative review summarizes and critically appraises the current evidence on genetics and the specific challenges of each type of AIS. The authors of the present study conducted an extensive review of numerous published articles and systematic studies on this topic. Using appropriate keywords or combinations thereof (androgen receptor, Morris syndrome and ambiguous genitals), the PubMed database was accessed, and English-language publications were selected.
Results
Genetic anomalies in androgen insensitivity syndrome
According to the literature, an AR gene mutation can be identified in > 95% of complete AIS (CAIS) cases, out of which approximately two thirds of these mutations are inherited through the maternal line, while the remaining one third are de novo mutations [8]. The AR gene is located in the Xq11-13 region of the X chromosome and contains 8 exons that encode the androgen receptor [8–10]. The AR is a large polypeptide (containing 920 amino acids) that belongs to the steroid hormone receptor family and it presents 4 functional domains (Table I) [11–19].
Table I
Functional domains of the androgen receptor gene
| N-terminal domain (NTD) |
|
| DNA-binding domain (DBD) |
|
| Hinge region | |
| C-terminal ligand binding domain (LBD) |
|
Fetal development genetics
The differentiation of the human embryo into the male sex is regulated by the sex-determining region Y gene (SRY), which is located on Yp 11.2. SRY encodes the testes determining factor (TDF), which has a crucial role in the initiation of male sex determination. This protein (TDF) is responsible for activation of two other proteins, SOX9 and SF1 (steroid genetic factor 1), which promote differentiation of the bipotent cells of the embryonic gonads into Sertoli cells, therefore leading to the appearance of the primordial testes. These two proteins (SOX9 and SF1) regulate the Sertoli cells’ production of anti-Mullerian hormone (AMH), which suppresses the development of the uterus and fallopian tubes from the Mullerian ducts, favouring instead the development of the Wolffian ducts. Leydig cells maturation is promoted by the Desert Hedgehog protein (DHH), which is produced by Sertoli cells. It has been reported that anomalies of this protein can be responsible for 46XY gonadal abnormal development [20–26]. Another factor involved in the male sexual differentiation process is fibroblast growth factor 9 (FGF), which is activated by the SOX9 protein. The absence of FGF9 can be responsible for the reversal of male to female phenotype [27].
In 46XX embryos, the absence of the Y chromosome explains the differentiation of the primordial bipotent embryonic gonad cells into ovaries, as well as the development of the fallopian tubes, uterus, cervix and the proximal part of the vagina from the Mullerian ducts. The absence of the SRY gene and TDF will determine the atrophy of the Wolffian ducts. In the absence of androgens, the clitoris will develop from the genital tubercle and the labia from the labial-scrotal folds [8].
In 46XY embryos with AIS, the activity of the SRY gene and TDF protein is normal, favours the development of the male gonads and inhibits the development of the uterus, fallopian tubes, cervix and upper part of the vagina via AMH action. During the intrauterine life, the normal virilisation process usually takes place between the 8th and the 14th week [28]. This process depends on androgen production and AR capability to bind the androgens. Therefore, the existence of a mutation that affects the androgen production or the androgen receptor will impair the normal virilisation process in 46XY embryos. Due to the insensitivity of androgen receptors to testosterone and dihydrotestosterone, AIS embryos will develop different phenotypes according to AR sensitivity to androgens, this varying from a complete feminine phenotype (in complete androgen insensitivity) to a male phenotype, but with altered spermatogenesis (mild androgen insensitivity). There is an intermediate form, characterized by partial androgen insensitivity, which leads to a male phenotype (usually these patients have associated malformations such as micropenis, hypospadias, bifid scrotum) or to ambiguous genitalia [29].
Type of androgen insensitivity syndrome
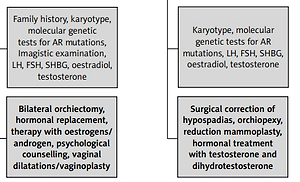
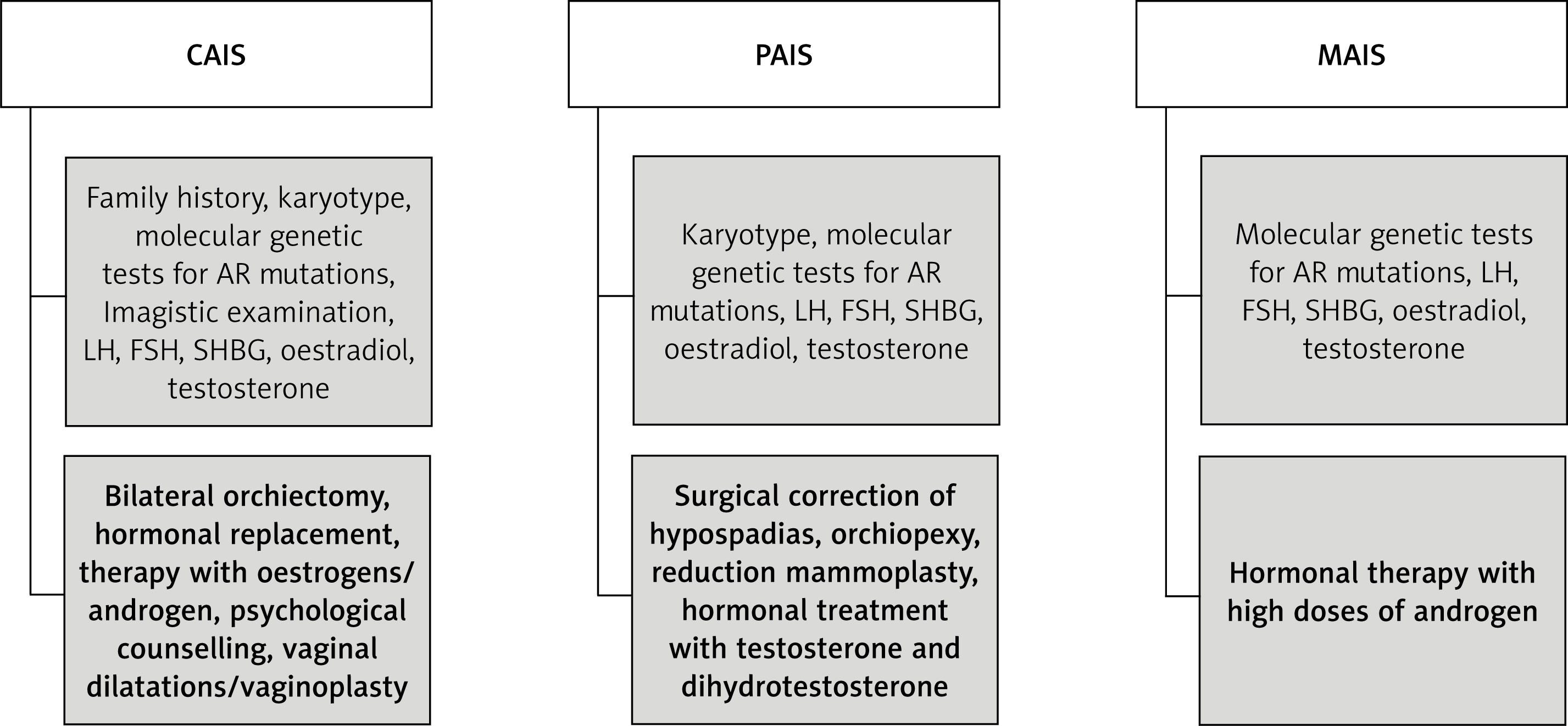
Considering the degree of AR sensitivity to androgen stimulation, this condition can be divided into three different entities: CAIS, partial AIS (PAIS) and mild AIS (MAIS). Based on personal experience and the review of the literature, we have summarised their clinical features, diagnostic and treatment pathways of management in Table II and Figure 1.
Table II
Type of androgen insensitivity syndrome and phenotype characteristics
Complete androgen insensitivity syndrome
Association of 46XY karyotype with an external feminine phenotype characterizes CAIS. The undescended testes are usually located in the lower abdomen, but they can also be found at any point along their descending path towards the scrotum (inguinal canals or labia majora). The internal genital organs are absent, as the result of the anti-Mullerian hormone produced by the testes. In almost all cases, the vagina has a shorter length compared with normal female patients and always presents a blind ending. Pubic and axillary hair is usually present in limited amounts, but it can also be completely absent, as a result of androgen insensitivity [1, 8]. CAIS patients also seem to have a lower average incidence of facial acne during puberty [10, 30]. The oestrogen level is similar to that found in the normal male; several researchers suggest that breast development is the result of cellular unresponsiveness to androgen stimulation, rather than to higher oestrogen production [1]. Another characteristic of these patients is the fact that they are taller than normal females, but shorter than men. This characteristic is related to the presence of a gene involved in the growth process, which is located on the Y chromosome (growth controlling gene – GCY) [1, 31].
Usually, these patients seek medical examination for primary amenorrhoea. In numerous cases, the suspicion of CAIS is raised during puberty, which usually occurs spontaneously. Puberty onset is debated among researchers, some of them reporting that puberty in CAIS patients is initiated in a later stage compared with female patients with 46XX karyotype; however, this hypothesis is not sufficiently supported. Most researchers consider that the moment of puberty onset in CAIS patients is similar to normal female patients [30]. Another scenario that could raise the suspicion of CAIS is the presence of an inguinal hernia or a labial swelling (the testes being located in the labia) in apparently normal female patients during childhood. The presence of a bilateral inguinal hernia in infant female patients is extremely rare. This finding, along with the presence of a short vaginal length, should raise the suspicion of CAIS.
In order to confirm the diagnosis, a karyotype assessment should be considered. Imaging investigations, readily available, reveal the absence of the uterus, fallopian tubes and ovaries and the presence of testes. Other elements useful for the diagnosis are family history (the existence of an older sister who has undergone surgery for inguinal hernia repair during childhood), incidental finding of the testes during imaging investigations and the existence of a mismatch between the karyotype and ultrasound findings during fetal life [11].
The oestrogen resulting from the aromatization process of the testosterone and the luteinising hormone (LH) are two key elements involved in puberty onset. In patients with CAIS, serum concentrations of LH are slightly above the upper values of those observed in men. The values of the follicle stimulating hormone (FSH) and sexual hormone binding globulin (SHBG) are also in the normal range [12]. In terms of oestradiol levels, its values are usually normal if we consider the male range, but slightly lower than those encountered in female patients. The testosterone levels are above the normal female range, but within the normal male values [13].
The presence of a feminine phenotype at birth, without any signs of external genital organs’ virilisation and without secondary male characteristics, is responsible for the fact that these patients are raised for long periods of time as females, having a female gender and identity, in other words considering themselves as being females and having typical feminine activities and interests [8].
After establishing the diagnosis of CAIS, the management of these patients involves bilateral orchiectomy, followed by hormonal replacement therapy with oestrogens, in order to avoid hypo-oestrogens symptoms, as well as psychological counselling. In some cases, vaginal dilatation and even enlargement vaginoplasty may be needed [1]. Bilateral orchiectomy is recommended because undescended testes are associated with a high risk of malignant degeneration after puberty. This risk is very small before adulthood and this allows the recommendation for bilateral orchiectomy to be postponed. Testicular preservation during childhood and adolescence favours normal puberty onset (with breast development and secondary sexual characteristics) under the action of oestrogens that results from androgen aromatization. If orchiectomy is performed before puberty, hormonal replacement therapy is mandatory in order to induce normal puberty [8]. In CAIS infants who have undergone bilateral orchiectomy, oestrogen substitution using either oral or transdermal oestrogen should be initiated at the age of 11–12 years, to induce normal pubertal onset with its specific changes and to obtain complete feminization. The treatment should start with a dose of 2 µg of ethinyl-oestradiol administered daily and it should be adjusted periodically over the next 2–3 years, in order to achieve a total daily dose of 30 µg, which is the normal adult dose [1, 2]. Oestrogen substitution therapy should be continued with a constant daily dose after achieving definitive breast development. A definitive oestrogen dose for the initiation of hormonal replacement therapy has not been defined. Some authors report a higher initiation oestrogen dose, in the range of 2.5–5 µg/day or 50–100 ng/kg daily oestrogen, which will be periodically increased until achieving a total daily dose of approximately 20–25 µg. Excessive doses of oestrogen may have a negative impact on the growth process by too early closure of the epiphysis [13, 32, 33].
Dehydroepiandrosterone is the most abundant circulating sex steroid hormone. Although it has no receptors to bind to, restoring the circulating levels of androgen hormones after orchiectomy was found to improve the sexual well-being. This is a clinical observation in CAIS patients after orchiectomy; the mechanism is not fully understood, but it may be explained by the effect of these hormones on brain and sexual development, despite the lack of receptors or receptor mutations.
Partial androgen insensitivity syndrome
Partial androgen insensitivity syndrome is the result of an incomplete cellular response to androgen stimulation. The phenotype of these patients depends on the degree of androgen receptors’ responsiveness to androgen stimulation. Most patients are born with incompletely developed and atypical male genitalia, or ambiguous genitalia at birth. Therefore, establishing the correct gender of new-born patients with genital anomalies and development of an appropriate therapeutic protocol require identification of the presence of an androgen receptor mutation for the diagnosis. Several studies suggest short periods of androgen administration in order to assess the AR response [2, 34].
In terms of hormone profile, these patients usually present high levels of luteinizing hormone and testosterone. In order to exclude the presence of an androgen synthesis defect, testosterone precursors, testosterone and dihydrotestosterone concentrations can be assessed, before and after human chorionic gonadotropin [8].
Most PAIS patients are raised as males. In borderline patients, with ambiguous genitalia, gender assignment should be given as soon as possible, considering the aspect of the genital organs. It is presumed that the more masculinized the genital organs are, the more likely it is that the brain has been virilised [35, 36]. The management of PAIS patients raised as males involves the surgical correction of hypospadias, orchiopexy, hormonal treatment with testosterone and dihydrotestosterone to induce puberty and to strengthen the virilisation process (it may improve penile length and other secondary sexual characteristics). The results of the androgen treatment can be seen after at least 6 months of treatment. Additional administration of androgen hormones is not always necessary, especially in cases with normal testes and normal testosterone production. For those patients who develop gynecomastia, reduction mammoplasty is necessary. The incidence of breast cancer in PAIS is low [2].
Mild androgen insensitivity syndrome
Mild androgen insensitivity syndrome is associated with a small degree of androgen insensitivity; these patients present a male phenotype. This type of androgen insensitivity is also the result of an androgen receptor mutation, but these patients, in contrast to those with PAIS and CAIS, present normally developed male genitalia. Until now, a small number of androgen receptor mutations that are strictly responsible for MAIS have been reported [1].
Usually, MAIS patients will seek medical advice for infertility, this being the main clue that could lead to the diagnosis. The product of serum testosterone and LH concentrations, used as an index of possible mild AIS in infertile men, could be considered a screening test for the presence of any mutation in the androgen receptor gene. Experimental results (performed in mice) of androgen receptor conditional clearance demonstrate that a functional receptor, expressed in Sertoli and Leydig cells, is essential for normal spermatogenesis [37]. Mild androgen insensitivity syndrome patients can also develop gynecomastia and reduced hairiness. High dose androgen treatment could restore these patients’ fertility [1].
Mild androgen insensitivity syndrome can also present as Kennedy’s disease, which is characterized by bulbar and spinal muscular atrophy, facial area, dysphagia, speaking difficulties, gynecomastia and infertility [38]. This disease is the result of an AR mutation, consisting in a hyper-expansion of the CAG repeat at the level of exon 1 [2]. Experimental tests in which transgenic mice (with hyper-expanded CAG repeats) were crossed with mice with inactive androgen receptors revealed increased neuromuscular and endocrine reproductive characteristics [39].
Based on the literature review and the authors’ personal experience in this field, we recommend the algorithm shown in Figure 1 for diagnosis and treatment of CAIS, PAIS and MAIS entities.
Androgen insensitivity syndrome and testicular cancer
In the case of AIS patients, the risk for germ cell cancer in gonads varies across the subtypes. It is considered that the number of germ cell tumours increases in individuals, simultaneous with AIS, because of the Y chromosome and presence of the testis-specific protein Y-linked 1 (TSPY) gene [40, 41]. The literature in the field is not consensual, showing variation of the risks of malignancy in the case of AIS [42–44].
The estimated risk of benign testicular tumours in patients with undescended testes during adulthood is approximately 25%, whereas the risk for malignant testicular tumours is lower, being estimated between 3% and 10% [45]. This risk is negligible during childhood and adolescence (< 1%) in CAIS patients, but it increases with age, being estimated < 3.5% at 25 years, and as high as 33% at 50 years [46]. Testicular germ cell tumours (TGCT) are preceded by in situ non-invasive lesions, known as germ cell neoplasia in situ (GCNIS). GCNIS has a 50% chance of becoming invasive over the following 5 years in the general population. The germ cell tumours associated with AIS correspond to type II seminoma, non-seminoma and dysgerminoma. In terms of benign tumours, the most frequently encountered are hamartomas and Sertoli cell adenoma. Carcinoma in situ originates from the primordial germ cells and it displays a large amount of germ cells in the seminiferous tubules [11, 47].
The low risk of neoplastic degeneration before adulthood allows orchiectomy to be postponed until after puberty. In spite of the risk of malignant degeneration, there are CAIS women who refuse orchiectomy. In such cases, periodic magnetic resonance imaging should be recommended, but this imaging investigation fails to detect germ cell in situ neoplasia [48].
Another recommendation is that the testes should be re-sited in the inguinal canals or at least in their proximity (to ensure a better follow-up) and a testicular biopsy should be performed during the procedure. Several tumour markers can be used to detect the pre-malignant changes characteristic for the carcinoma in situ (PLAP: placental like alkaline phosphatase, SCF: stem cell factor, OCT3/4: octamer binding transcription factor 3/4), but these markers can be used only on testicular tissue samples obtained after biopsy [49–51].
In patients with PAIS and undescended testes, the incidence of testicular malignant degeneration is significantly higher compared to CAIS patients, up to 50%. Therefore, in order to avoid this risk, scrotal orchidopexy should be recommended in all PAIS male patients and laparoscopic orchiectomy in female PAIS patients [52, 53].
Androgen insensitivity syndrome and prostate cancer/benign prostatic hyperplasia
In patients with CAIS, prostate development and masculinization are inhibited by the impaired androgen receptor function, the prostate remaining practically undeveloped.
In PAIS patients, prostate is significantly smaller compared to normal male patients, often being impalpable, whereas MAIS patients usually present a quasi-normally developed prostate. Therefore, the risk of prostate cancer and benign prostatic hyperplasia in patients with CAIS is virtually absent, whereas in patients with partial and mild androgen resistance syndrome this risk can be increased by the exogenous androgen supplementation with testosterone and dihydrotestosterone, as well as by the type of androgen receptor mutation.
Salmasi et al. assessed the PSA levels in 26 patients diagnosed with 46XY disorders of sexual development (including three CAIS cases and nine PAIS cases). These authors reported that 18 patients presented PSA levels lower than 0.1 ng/ml, half of them being raised as females. The remaining 9 patients presented PSA levels ranging between 0.1 and 0.9 ng/ml (all of them being raised as males), which were in the same range as those found in normal male patients with similar age and race; therefore these patients could be associated with the same risk of prostate cancer as normal male patients. The authors mentioned that patients raised as females did not present detectable PSA levels and recommended that the screening for prostate cancer using PSA and digital rectal examination in such patients (including PAIS and MAIS cases) should be done as in normal male patients [54].
Discussion
Among 46 XY disorders in the sex development, AIS is known and recognised as the most common type, characterised by a vast field of clinical heterogeneity [55]; also, it is caused by abnormalities in the AR gene, with phenotypes that vary from CAIS (having a completely female phenotype) to PAIS (having a low degree of infertility or under virilisation) [56]. Each type of AIS presents a series of specific challenges. In the CAIS patient, after bilateral orchiectomy is performed, oestrogen levels will decline. These patients present a high risk of developing osteoporosis, cognitive decline and cardiovascular disease if the oestrogen deficit is not compensated [13, 57]. In CAIS patients who undergo bilateral orchiectomy after puberty, as well as in those with orchiectomy during childhood and oestrogen substitution therapy during puberty, hormonal replacement therapy consists in a dose of 1 mg of oestradiol administered daily or 40–50 µg administered transdermally. The transdermal approach seems to provide a more physiological reaction [11].
A considerable percentage of CAIS patients who undergo orchiectomy and hormonal oestrogen substitution will complain of a decrease in their general well-being, as well as about sexual dissatisfaction. Numerous patients report a significant decrease of arousal, libido, orgasm incidence and orgasm intensity after gonadectomy. It seems that testosterone administered after orchiectomy in CAIS patients will improve these complaints. Birnbaum compared the effectiveness of androgen substitution therapy in 26 CAIS patients following orchiectomy with the usually prescribed oestrogen hormonal replacement treatment. The authors noted that the two treatment options (androgens and oestrogens) did not differ in terms of their psychological and mental well-being effects, or in their impact on quality of life [58]. Nevertheless, androgen substitution therapy seems to be superior when compared to oestrogen treatment in terms of sexual well-being and orgasmic function [59].
For the PAIS patients assigned and raised as females, the therapeutic protocol implies bilateral orchiectomy during childhood, to prevent the effects of further virilisation and to reduce the risk of testicular cancer, in patients with undescended testes. Further surgery, such as genital reconstruction, is usually needed, to achieve a more natural appearance of the genital organs and also for improving their functionality and the patient’s well-being. The success of such procedures significantly improves the patient’s quality of life [31]. Hormonal treatment with oestrogen is mandatory, to induce puberty and ensure the proper development of secondary female sexual characteristics. Compared with CAIS patients, PAIS patients are more susceptible to developing anxiety, depression and other psychiatric disorders [60].
In the case of MAIS, information on clinical results is scarce. Infertility and gynecomastia are the usual way of presenting this phenotype [61], mastectomy being the recommended procedure to correct gynecomastia. This phenotype can be observed and found in people diagnosed with Kennedy disease [9].
Psychosexual identification is a major challenge in AIS management. It is considered that the sexual differentiation not only consists in genitalia formation, but also in brain effects of androgens. Psychosexual development is a complex process, many factors being involved, such as genetic influence, hormones, environment, social life, etc. [62, 63]; it includes gender identity, self-identification to a gender and sexual orientation, defined as the response of a person to sexual stimuli. Both can be affected in AIS, because of misalignment between chromosomal, gonadal and phenotypic sex [62].
Patients with CAIS should be raised as females, because of external genitalia development and androgen unresponsiveness of the brain. These patients require psychological advice after the diagnosis is established and regarding the necessity of orchiectomy and acceptance of infertility [64].
Patients with PAIS are raised as males; the female gender is preferred only in cases with severe PAIS. They experience more often psychological distress, 25% of them suffering from a sexual identity crisis. Psychological support is needed, and a decision of assigned gender is made by the family and medical team [64, 65].
In conclusion, androgen insensitivity syndrome is a frequent 46XY disorder of sex development, the mechanism involved being an AR mutation. The clinical presentation of AIS depends on the AR sensitivity to testosterone and dihydrotestosterone stimulation, varying from male phenotype and infertility to female phenotype with a 46XY karyotype. The diagnosis in patients with mild AIS is made usually during adulthood due to infertility or during puberty due to the development of gynecomastia, whereas in patients with complete androgen insensitivity the diagnosis can be made during pregnancy, at birth, during childhood or adulthood. For cases with PAIS, the diagnosis is made in most cases at birth due to the presence of ambiguous genitalia. The treatment in CAIS patients implies bilateral orchiectomy to prevent malignant degeneration of the testes and hormonal therapy to maintain the normal female phenotype, despite the 46XY karyotype. The timing of gonadectomy has been debated by many authors, but current guidelines recommend postponing it until after puberty, to achieve complete natural feminization and due to the fact that the risk of testicular tumours in CAIS patients is low before adulthood. Compared with CAIS patients, PAIS patients present a higher risk of developing testicular tumours. Establishing the correct sex assignment may be very difficult in PAIS patients, because they present ambiguous genitalia. The management of AIS requires a multidisciplinary team of paediatricians, endocrinologists, gynaecologists, urologists, plastic surgeons and psychiatrists. Psychological counselling should be recommended for these patients and their parents.