Introduction
Bilirubin is an oxidative end product of heme catabolism and is excreted by liver after glucuronidation by hepatic uridine diphosphate glucuronosyltransferase [1]. Hyperbilirubinaemia remains one of the most frequent clinical diagnoses in the neonatal period, and more than 60% of newborns develop clinical jaundice in the first week of life [2, 3]. Severe hyperbilirubinaemia may progress to acute kernicterus or bilirubin encephalopathy with a significant risk of mortality in newborns [4–6]. Moreover, recently hyperbilirubinaemia has emerged as a significant risk factor for non-syndromic auditory neuropathy due to dys-synchrony in the auditory nerve or brainstem auditory pathways, causing auditory processing problems or neural, not sensory, hearing loss [7].
Total bilirubin is composed of conjugated and unconjugated bilirubin [8]. Unconjugated bilirubin is a potent neurotoxin, which, by crossing the blood‒brain barrier, may injure neural cells. Once in the brain, bilirubin has been reported to lead to impaired myelination, astrocyte and microglia activation, and neuronal cell death [9]. Except for the traditional recognised areas of fulminant injury to the globus pallidus as observed in infants with kernicterus, the hippocampus, one of the important regions of brain for memory and learning function [10], is a major target of bilirubin in the brain [11].
A previous study has indicated that exposure of hippocampal slice cultures to unconjugated bilirubin leads to long-term depression (LTD) induction and an impairment of CA1 long-term potentiation (LTP) in a time- and concentration-dependent manner [1]. LTP is a form of synaptic plasticity that is believed to form the cellular basis for memory and learning [12]. It was suggested that such deficits in LTP may underlie some cognitive impairments associated with unconjugated bilirubin exposure. Therefore, investigations of hyperbilirubinaemia-induced impairment of synaptic plasticity are of great importance to the mechanism underlying the effects of hyperbilirubinaemia on cognitive impairment.
Herein we aim to investigate the effect of hyperbilirubinaemia on synaptic plasticity in the dentate gyrus (DG) region of hippocampal rats. Seven-day-old healthy SD rats were randomly divided into a control group and an experiment group. The I/O functions, paired-pulse ratio (PPR), excitatory postsynaptic potential (EPSP), and population spike (PS) amplitude were measured in the DG area of the two groups of rats in response to stimulation applied to the lateral perforant path.
Material and methods
Experimental animals and treatment
Healthy Sprague Dawley rat pups (seven days old) were used in the study. According to Vannucci et al. [13], the development of the brain of a rat at this age is histologically similar to that of a 32- to 34-week gestation human newborn infant. Forty female rats (15.6–19.4 g) were obtained from the Experimental Animal Centre of Anhui Province (Animal license: SCXK, 2005–001). The rats were fasted for 12 h and divided into two groups (n = 20 in each group). The first group was injected (i.p.) with bilirubin solution. Bilirubin solution was obtained from Sigma (St Louis, MO) and purified according to the method of McDonagh [14]. Bilirubin was dissolved in 0.5 M NaOH and was prepared immediately before use under light protection to avoid photodegradation; the pH was restored to 8.5 by addition of 0.5 M HCl. The study was approved by AnHui Medical University Ethics Committee. Rats in the experiment group were injected (i.p.) with 0.342 M bilirubin solution based on the previous study [15]. The other 20 normal rats were injected with normal saline and used as the control group. The rat model of hyperbilirubinaemia was validated after 48 h; in our study, 16 rats were identified as the successful model of hyperbilirubinemia. At the age of 80–100 days, the animals were utilised for extracellular recording in the area of the DG of the hippocampus.
Stimulation and recording
Rats were anaesthetised with urethane (1.5 g/kg, i.p.), and the head was fixed in a stereotaxic head-holder (under deep ether anaesthesia). A concentric bipolar stimulating electrode (0.25 mm outer diameter, 0.75 mm tip separation; Rhodes Medical Instruments, Woodland Hills, CA) was placed in the lateral perforant path. A glass micropipette recording electrode (3–5 µm tip diameter, 1–3 MΩ, co-ordinates: 3.8 mm posterior to bregma, 2.5 mm lateral to the midline) was lowered into the DG until the maximal response was observed (3.0–3.5 mm ventral). Glass micropipettes filled with 2 M NaCl were used for extracellular recordings.
I/O function
I/O curves were generated by systematic variation of the stimulus current (0.1–1.0 mA) in order to evaluate synaptic potency as previously described [16]. Stimulus pulses were delivered at 0.125 Hz.
Paired-pulse ratio
The paired-pulse ratio (PPR) was evaluated by increasing the inter-pulse intervals (IPIs, 10–400 ms) as previously described [16]. The stimulus current intensity was adjusted at an intensity yielding about 50% of the maximal amplitude of the population spike (PS). Stimulus pairs were delivered at 0.05 Hz.
Long-term potentiation
Long-term potentiation were recorded as previously described [16]. The stimulus intensity selected for baseline measurements was adjusted to yield about 40% of its maximal amplitude. Baseline recordings were obtained for 30 min at 8-s intervals (0.125 Hz), followed by application of the high-frequency stimulation (HFS: 250 Hz, 1 s) and a post-tetanic recording period of 1 h with a single pulse.
Data analysis
All data were shown as the mean ± SEM. Data were recorded using an Igor Pro 6.2 (Wave Metrics, OR) and analysed with Origin 7.5 software (Origin Lab, MA). The PS amplitude and the EPSP slope was measured as previously described [12]. Two-way analysis of variance (ANOVA) followed by χ2 test was performed to compare the difference between the groups. P < 0.05 was considered as statistically significant.
Results
I/O functions
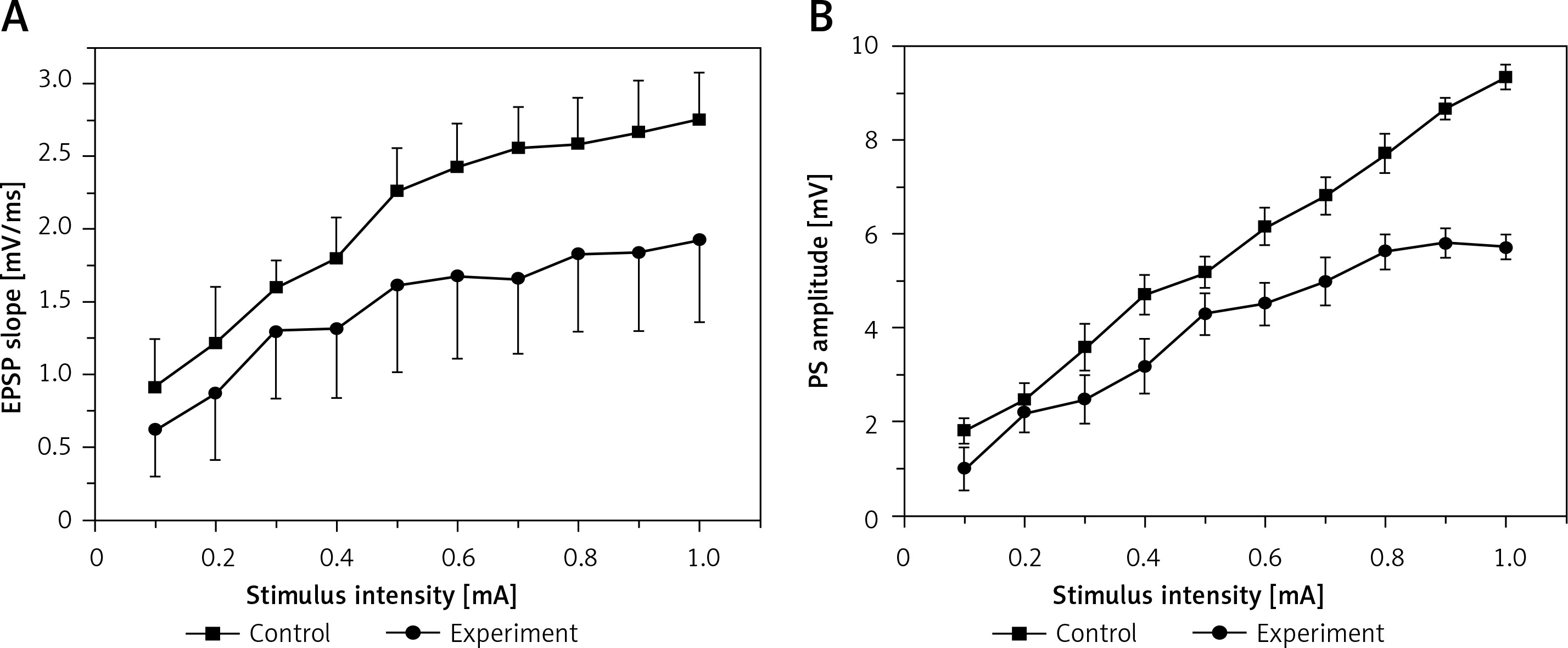
Figure 1 illustrates the I/O curves in the DG, which were measured as EPSP slope (A) and PS amplitude (B) before induction of LTP in control and hyperbilirubinaemia rats. Compared with those of the control rats, the current-voltage curves of both EPSP slope and PS amplitude in the experimental rats were significantly depressed (EPSP: F = 11.539, p < 0.01; PS: F = 102.224, p < 0.01). The result suggests that hyperbilirubinaemia significantly depressed the basic synaptic transmission.
Figure 1
Input/output (I/O) curves (mean ±SEM) of the excitatory postsynaptic potential (EPSP) slope (A) and population spike (PS) amplitude (B) in the dentate gyrus (DG) area in the control group and in the experimental group as a function of stimulus intensity before induction of long-term potentiation (LTP). Both EPSP slope (p < 0.01) and PS amplitude (p < 0.01) were significantly depressed in the experimental group compared with the control group
Paired-pulse reactions
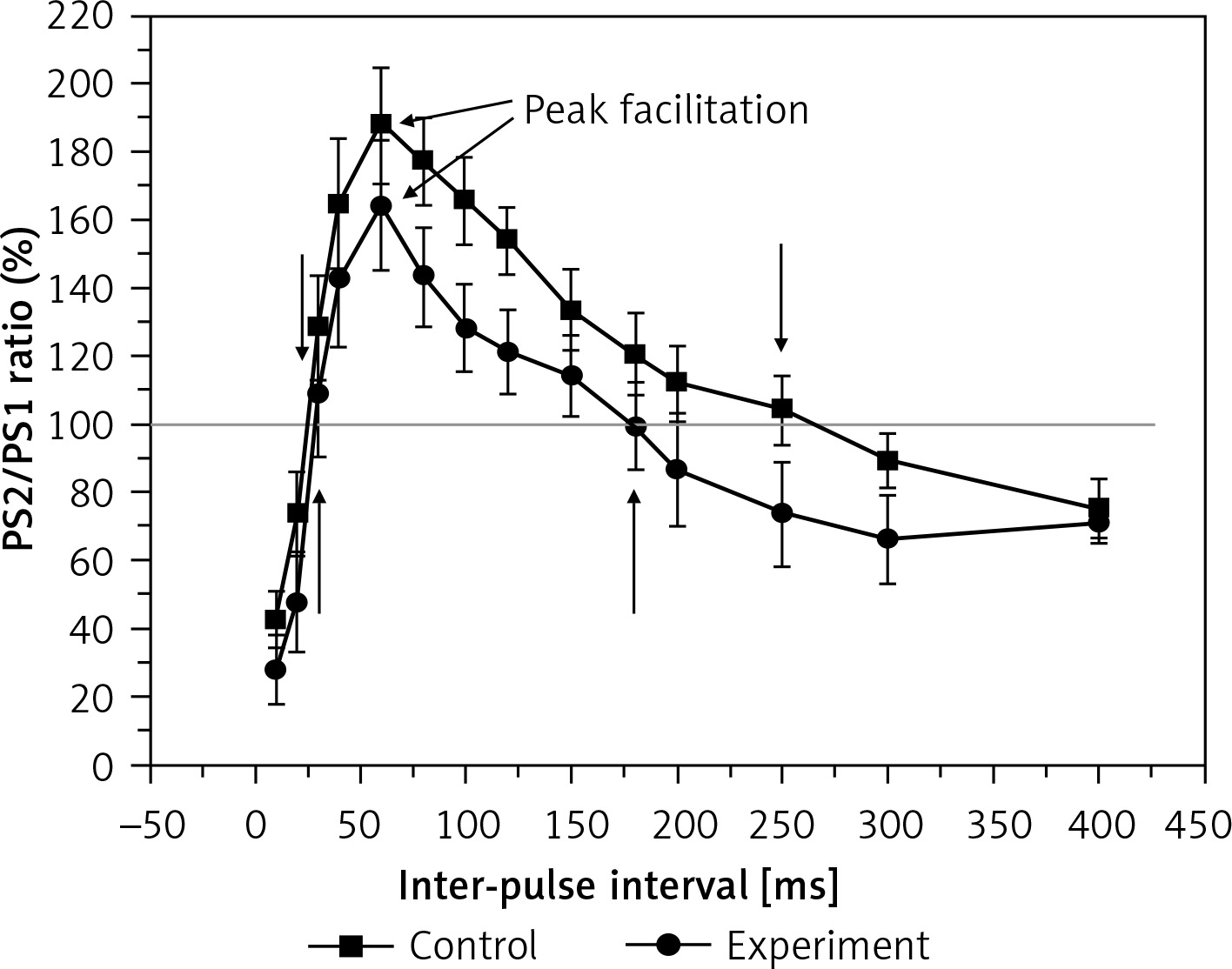
To examine the effect of hyperbilirubinaemia on the short-term synaptic plasticity of rats, we measured the PPR of the PS amplitude. The results in Figure 2 show the PPR of the PS amplitude in the control and experiment groups. The PS2/PS1 is defined as a PPR ratio to quantify the effect as enhancement or inhibition of the second response relative to the first. The peak facilitation (the values of the extreme point of the curve) and the facilitation period are used to describe the differences in the two groups. The average peak facilitation was 187 ±16% in the control and 164 ±18% in the experiment group (F = 21.054, p < 0.01). The facilitation period duration of PS was 155 ms in the experimental rats, which was less than that of the controls (235 ms). This result shows that hyperbilirubinaemia impairs paired-pulse facilitation (PPF) significantly.
Long-term potentiation
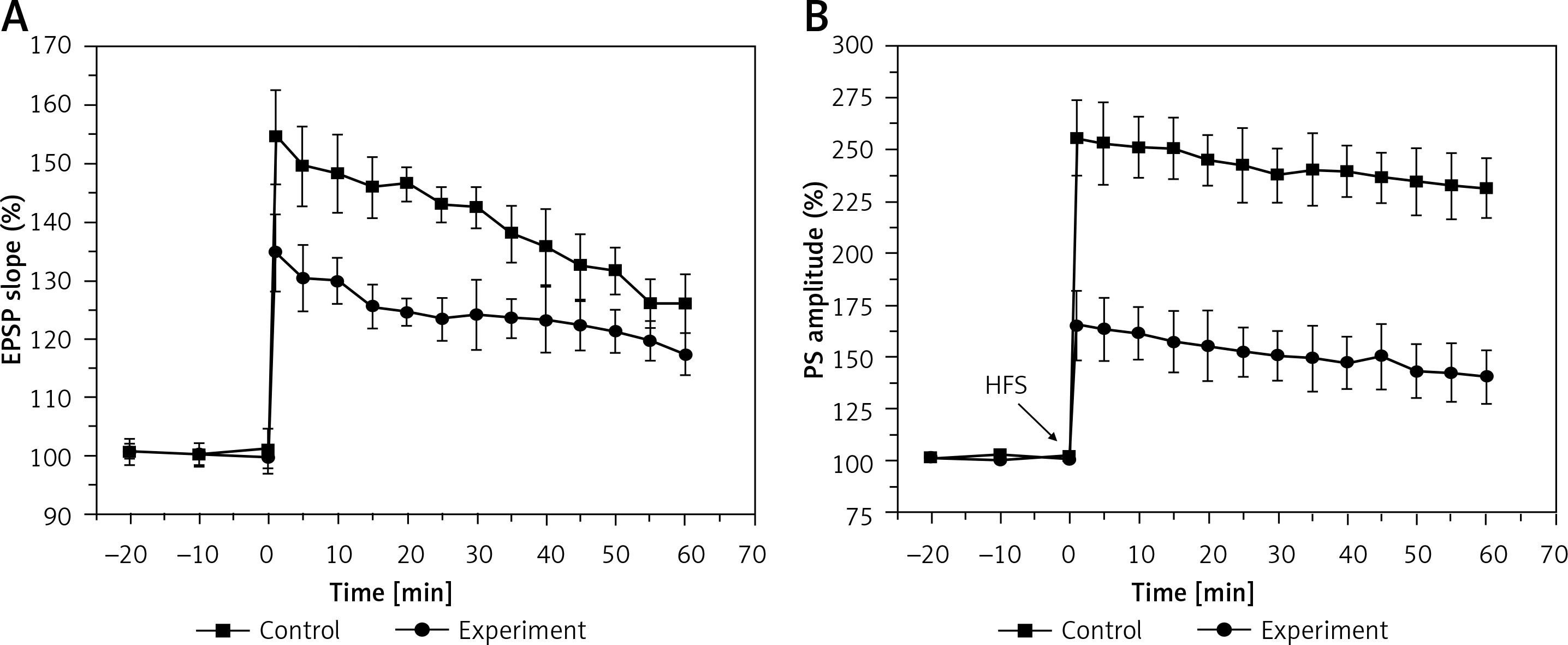
The results in Figure 3 show the amplitudes of LTP on both the EPSP slope (A) and the PS amplitude (B) in the control and experimental groups. In the control group, the LTP amplitudes were 140 ±3.5% and 242 ±6%, when estimated from the EPSP slope and PS amplitude, respectively, which were significantly depressed to 124 ±3.4% (EPSP slope, F = 70.489, p < 0.01) and 138 ±8.6% (PS amplitude, F = 253.46, p < 0.01) in the experiment group. These results demonstrated that hyperbilirubinaemia induced a deficit of LTP in the DG area of the hippocampus.
Figure 3
Long-term potentiation (LTP) of the excitatory postsynaptic potential (EPSP) slope (A) and population spike (PS) amplitude (B) in the control and experiment group. Both EPSP slope LTP (p < 0.01) and PS amplitude LTP (p < 0.01) were significantly depressed in the experiment group compared with those in the control group. Downward arrows indicate the time when the high-frequency stimulus (HFS) was applied
Discussion
This study suggests that hyperbilirubinaemia induces impairment of synaptic plasticity in the rat DG area in vivo, including PPR, I/O function, and LTP. I/O function reflects the basal synaptic response, which can reflect not only the postsynaptic receptor response, but also the level of presynaptic neurotransmitter release [12]. PPR is a short-term synaptic plasticity [17] and is a facilitation of a second response when a synapse is stimulated twice with a short interval (20–400 ms) [12]. LTP is an electrophysiological model of activity-dependent plasticity that embodies the cellular components of information storage at the synaptic level, and is widely accepted as a primary mechanism of memory [18, 19]. Previous novel findings also showed that short-term exposure to bilirubin can inhibit the induction of LTPs in the hippocampus [20]. It is well-accepted that memory is intimately associated with the plasticity of neuronal synapses in the hippocampus and a decline in cognitive function directly correlates with loss of synapses [21]. Thus, our study may indicate that hyperbilirubinaemia has an effect on cognitive impairment.
Many clinical investigations have suggested a close association between high total serum bilirubin levels and the neurodevelopmental risks in newborns [22–24]. A recent study reported the 30-year results of a prospective follow-up study on a birth cohort of full-term and normal-weight newborns with hyperbilirubinaemia, and concluded that 45% of the cases identified with neonatal hyperbilirubinaemia showed at least one neurobehavioral disability at the age of 9 years (significantly higher than rates in controls), and these difficulties appeared to continue into adulthood [25]. Also, an earlier study suggested that that prolonged exposure of clinically relevant concentrations of unconjugated bilirubin exposure impairs the induction of hippocampal CA1 LTP and LTD in rat organotypic slice cultures [1]. Hansen et al. [26] indicated that the effect of bilirubin on rat hippocampal slices consists of a gradual and reversible decrease in synaptic transmission by causing the presynaptic fibre volley (PV)/EPSP curve to shift to the right while the EPSP/population spike (PS) curve shifted to the left.
The molecular mechanism was explored in several studies. Li et al. reported that bilirubin facilitates presynaptic glutamate release and enhances glutamatergic synaptic transmission by activating postsynaptic AMPA and NMDA receptors, which results in neuronal hyperexcitation [27]. Another study indicated that the increased intracellular calcium concentration [Ca2+]i, the perturbations of plasma, mitochondrial, and/or endoplasmic reticulum membranes or mitochondrial energy failure are likely to be linked temporally and spatially in the pathogenesis of bilirubin induced neuronal injury [28]. Moreover, a previous study summarised that low levels of free bilirubin may promote programmed neuronal death, corresponding to an apoptotic process that requires the participation of NMDA receptors and involves caspase activation, along with bilirubin-induced inhibition of protein kinase C activity [29]. Whether bilirubin-induced depression of synaptic transmission is due to apoptotic neurodegeneration requires further investigation.
In conclusion, these findings suggest that hyperbilirubinaemia could induce impairment of synaptic plasticity in the rat DG area in vivo, including I/O function, PPR, and LTP, which may be closely related to cognitive impairment.