
Introduction
Investigations into melanoma and other cancer types have suggested a potential role for dysregulated cholesterol homeostasis in the development of cancer [1–8]. Experimental studies have shown that a high-fat diet enhances tumour growth and metastasis of xenograft human melanoma cells [9, 10]. Cholesterol can activate cancer-related signalling pathway, e.g. the hedgehog pathway, through direct binding to the G-protein-coupled receptors, including smoothened receptor [11, 12] and adenosine A2A receptor [13], by which affect cell differentiation and proliferation as well as tumour formation [14]. Cholesterol can also activate other oncogenic signalling pathways through binding to the scaffold proteins such as NHERF1/EBP50 [15], which regulate oncogenic signalling networks via assembling cancer-associated proteins, including those involved in the Wnt/β-catenin and PI3K/Akt pathways [16].
Although disruption of cholesterol homeostasis has been proposed to contribute to cancer development, the potential of therapeutic targeting of cholesterol homeostasis is a debatable issue in oncology. Previous studies of lipid-lowering drugs have shown an association between cancer risk and low levels of low-density lipoprotein (LDL) cholesterol (LDL-C) [17]; however, extant evidence suggests that the drugs per se do not enhance cancer risk [18–21].
Proprotein convertase subtilisin/kexin 9 (PCSK9) is an important modulator of cholesterol haemostasis through controlling the clearance of the plasma LDL-C from the bloodstream via regulation of the liver’s LDL receptor (LDLR). PCSK9 was found to be a therapeutic target for lowering LDL-C since early studies showed that gain-of-function mutations in PCSK9 are causally associated with the elevated plasma level of LDL-C [22], while loss-of-function mutations are associated with hypocholesterolaemia and a decreased risk of coronary artery disease [23–26]. Although scientific evidence supports the benefits of PCSK9 inhibitors in lowering LDL-C and reducing cardiovascular outcomes [27–33], the lipid-independent effects of PCSK9 inhibition, especially in patients with cancer, has remained largely unexplored. Hitherto, there have been several human studies that evaluated a possible link between PCSK9 polymorphisms and the risk of cancer, and the results have been contradictory [34–36]. A previous study could statistically associate LDL-raising PCSK9 variants with a higher risk of the cancer [34], while another study could not validate an association between PCSK9 loss-of-function variants and increased cancer incidence [35]. In contrast to these reports, a recent Mendelian-randomisation study showed that PCSK9 LDL-raising variants are associated with a higher risk of cancer, while LDL-lowering polymorphisms mimicking PCSK9 inhibitors were reported to be associated with a reduced risk of cancer occurrence [36]. These inconsistent results necessitate additional studies to evaluate the efficacy and safety of PCSK9 inhibitors in cancer.
Anti-PCSK9 vaccines are emerging PCSK9 inhibitors in the pipeline, which have shown significant LDL-lowering effects in experimental models of hypercholesterolaemia [37–40]. We previously formulated a nanoliposomal anti-PCSK9 vaccine that could efficiently promote long-lasting, specific, and safe anti-PCSK9 antibodies in BALB/c mice. Nanoliposomal vaccine-induced antibodies were found to target plasma PCSK9 and suppress its binding to LDLR, thereby leading to the inhibition of PCSK9 function [41]. The present study was carried out to assess the effects of the mentioned nanoliposomal anti-PCSK9 vaccine in C57 BL/6 mice bearing B16F0 melanoma.
Material and methods
Vaccine preparation and characterisation
Preparation and characterisation of the liposome nanoparticles
The thin-film lipid hydration method was employed to manufacture nanoliposome formulation containing 1,2-Dimyristoyl-sn-glycero-3-phospho-rylcholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-phosphorylglycerol (DMPG), and cholesterol (Chol) (Avanti Polar Lipid; Alabaster, USA) at the final concentration of 40 mM (total phospholipids and Chol). In brief, DMPC, DMPG, and Chol were mixed in chloroform at the molar ratios of 75 : 10 : 15, respectively. Lipid mixture was dried to a thin lipid film under decreased pressure by rotary evaporation (Heidolph, Germany). Then, the obtained lipid-film was freeze-dried (VD-800F, Taitech, Japan) overnight to entirely eliminate the organic solvent. Afterward, the dried lipids were hydrated with 10 mM HEPES buffer (pH 7.2) containing 5% dextrose, and vortexed and bath-sonicated to disperse completely into the buffer. To obtain small unilamellar vesicles (SUVs) with a uniform size of 100 nm, the multilamellar vesicles (MLVs) were extruded using a mini extruder (Avestin, Canada) with polycarbonate membranes of 600, 400, 200, and 100 nm pore size, respectively. Particle size (diameter, nm), zeta potential (surface charge, mV), and poly dispersity index (PDI) of the prepared nanoliposomal formulation were determined using dynamic light scattering (DLS) technique on a Zetasizer (Nano-ZS,Malvern, UK) at room temperature (RT). The synthesised nanoliposomes were stored at 4°C under argon.
Construction of immunogenic peptide
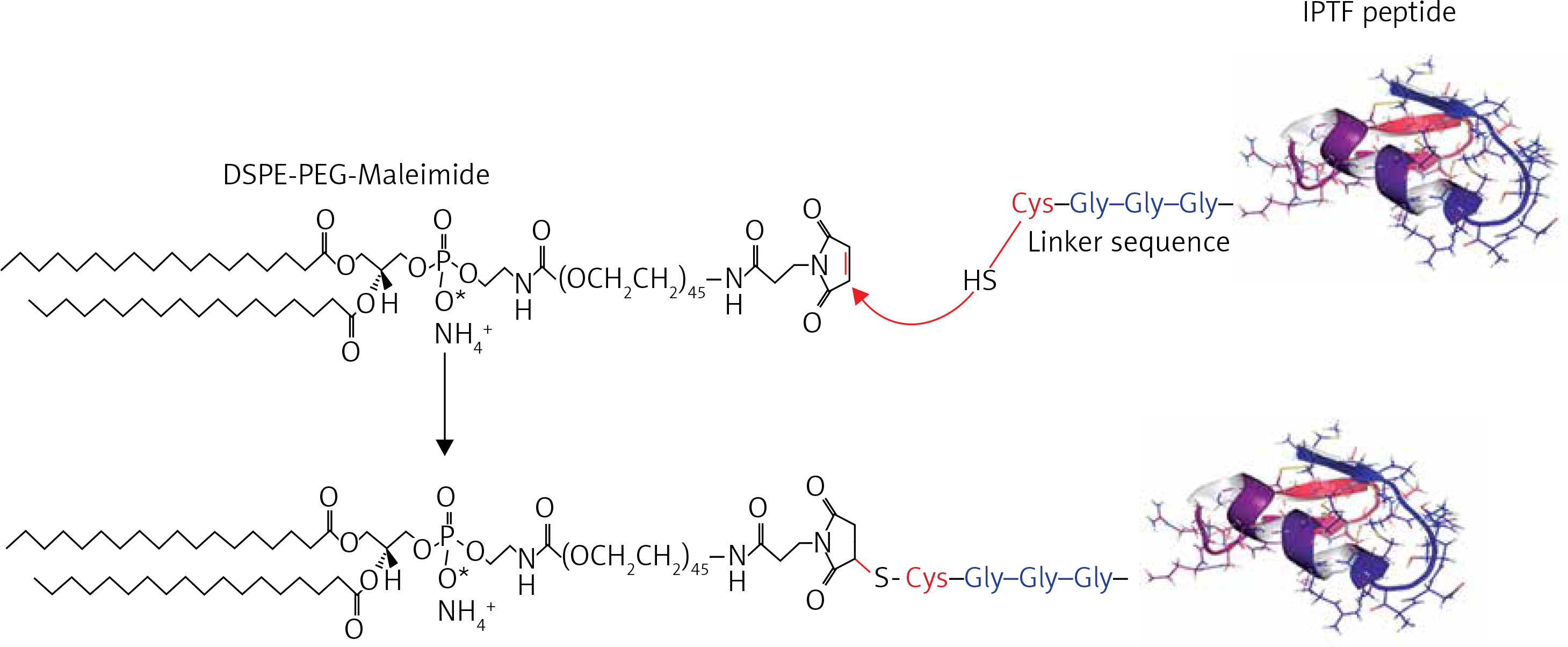
The immunogenic fused PCSK9-tetanus (IFPT) peptide with a purity grade of > 95% was synthesised and high-performance liquid chromatography (HPLC)-purified by ChinaPeptides Co., Ltd. (Shanghai, China). Already designed, the IFPT peptide [42] includes a PCSK9 sequence as a B cell epitope inspired from the AFFiRiS group [43, 44], and a T-helper cell epitope belonging to tetanus toxin used as a pharmaceutically acceptable adjuvant carrier [45] (Table I). To conjugate IFPT epitope on the surface of liposome nanoparticles, it was linked to DSPE-PEG-Mal (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(PEG)-2000]) lipid (Lipoid GmbH, Germany) via an N-terminal cysteine residue added to IFPT peptide (Figure 1).
Table I
Sequence of the immunogenic peptides used in the present study
| Peptide name | Sequence | Immunogenicity |
|---|---|---|
| PCSK9 | S-I-P-W-N-L-E-R-I-T-P-V-R | B cell epitope |
| Tetanus | A-Q-Y-I-K-A-N-S-K-F-I-G-I-T-E-L | T cell epitope |
| IFPT | *CGGGSIPWNLERITPVRAQYIKANSKFIGITEL |
Construction of DSPE-PEG-IFPT micelles
DSPE-PEG-Maleimide lipid was employed to attach the immunogenic peptides on the surface of liposome nanoparticles as an adjuvant delivery system. N-terminal cysteine residue of the IFPT peptide provides a thiol group that reacts with pyrrole group maleimide and produces a thioether bond; thereby, the peptide covalently links to DSPE-PEG-Maleimide lipid. The IFPT peptide and DSPE-PEG-Mal at the molar ratios of 1.2 : 1, respectively, were mixed in DMSO/chloroform solution at the volume ratio of 1 : 1 and then slowly stirred at RT for 48 h. The linkage was verified by TLC (thin layer chromatography) method with the mobile phase containing chloroform, methanol, and water at the volume ratio of 90 : 18 : 2. Then, the DMSO/chloroform solution was dried by rotary evaporator and freeze-drying followed by hydration with sterile deionised water (pH 7.2) at 30oC to construct DSPE-PEG-IFPT micelles. The efficiency of the linkage in the prepared micelles was determined by HPLC analysis. The true value of the linked micelles was determined using efficiency of linkage and content of total lipid measured by the Bartlett phosphate assay method [46].
HPLC analysis of the linkage efficiency
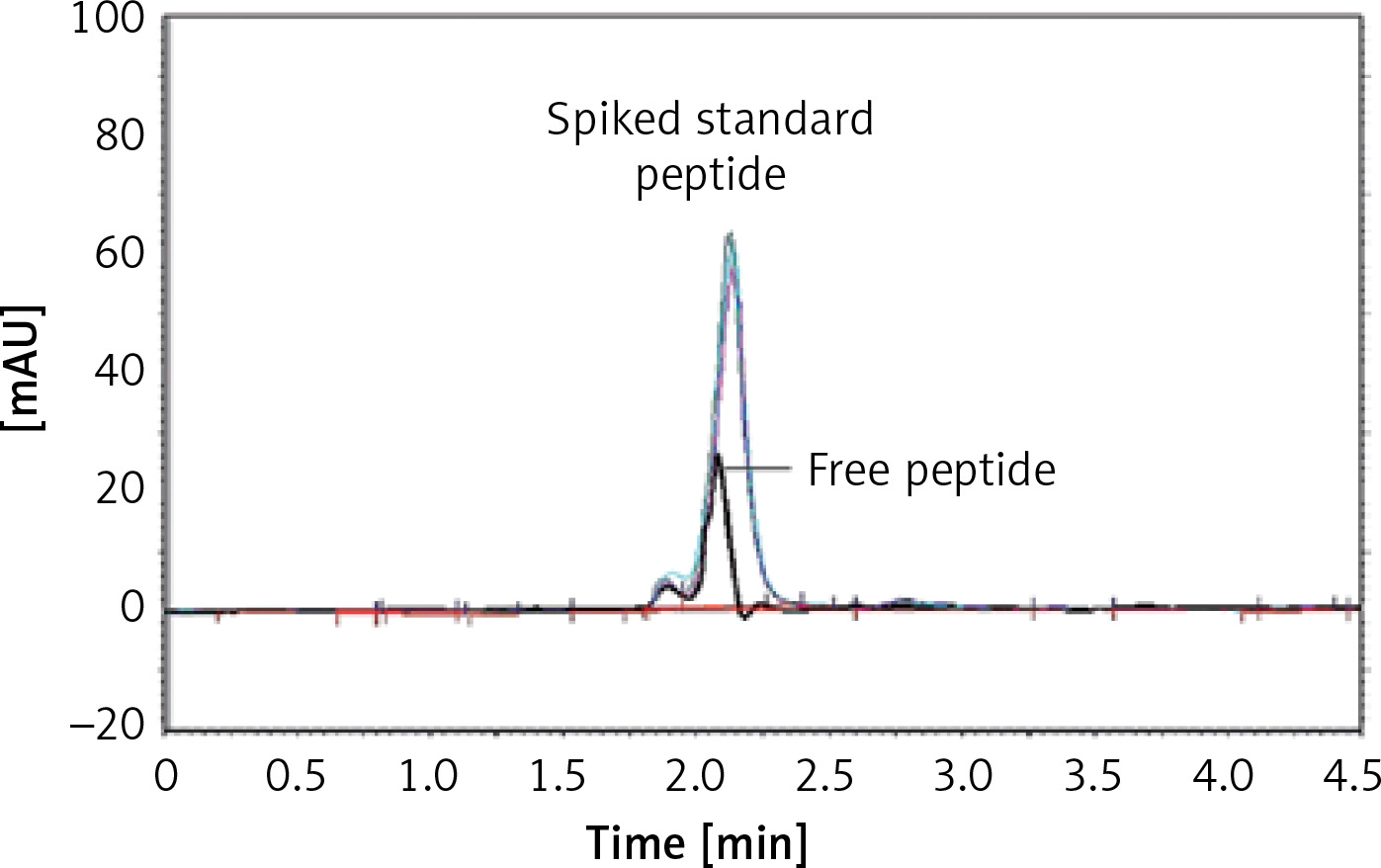
The efficiency of the linkage between the IFPT peptide and DSPE-PEG-Mal linker was indirectly measured by HPLC quantification of the free peptide content of the prepared DSPE-PEG-IFPT micelles. The HPLC apparatus was equipped with a Smart Line HPLC Pump 1000, a PDA Detector 2800 (set at 220 nm), and a Degasser5000, all from Knauer (Berlin, Germany). Each sample (20 μl) was injected through a Smart Line Auto Sampler, and data were obtained and processed with ChromGate software (version 3.3.1) from Knauer (Berlin, Germany). Chromatographic separation was performed on a C18 reverse-phase column, 4.6 mm × 25 cm (Shimadzu, Japan), using an isocratic mobile phase of (0.1% TFA in water)/(0.1% TFA in acetonitrile) at gradient ratios of 55/45 to 45/55 in 10 min, at a flow rate of 1 ml/min.
The IFPT peptide with HPLC purity > 95% was used as a standard solution. The free peptide peak within the chromatogram of the micelle sample was identified and assigned based on the retention time (2.2 min) of the standard solution, followed by sample spiking. To quantify the free peptide content of the micelle sample, a calibration curve was constructed by injecting standard solution at five concentrations (50–500 μg/ml), which was linear, with a correlation coefficient (r 2) of 0.9954. Using linear regression analysis of the calibration curve appearing in the standard chromatogram, the free peptide content of the micelle sample was estimated. Linkage efficiency in the constructed DSPE-PEG-IFPT micelles was calculated by subtracting the free peptide amount within the micelles quantified by HPLC from the amount of the IFPT peptide initially added.
Construction and characterisation of nanoliposomal IFPT vaccine
Nanoliposomes were used as a delivery adjuvant to enhance immunogenicity of the peptide. Because many IFPT peptides can be conjugated to the surface of liposome nanoparticles, we propose that IFPT-linked nanoliposomes can elicit a high-titre antibody against self-antigen PCSK9, maybe through elevating peptide valency. To conjugate the IFPT peptide on the nanoliposome surface, the post insertion approach was performed, in which the prepared DSPE-PEG-IFPT micelles (100 μg, based on the linked peptide) and liposome nanoparticles (1 ml) were mixed and then gently shaken at 45°C for 3 h. The micelles were inserted in the nanoliposome bilayer via DSPE phospholipid moiety, and expose IFPT peptides on the nanoliposome surface through the PEG chains. Particle size, surface charge, and PDI of the prepared nanoliposomal IFPT particles were evaluated using DLS technique on a Zetasizer (Nano-ZS, Malvern, UK) at RT. The IFPT-conjugated nanoliposomes were adsorbed to 0.4% Alum adjuvant (Sigma-Aldrich) at a 1 : 1 (v:v) ratio in a total volume of 400 μl and stored at 4°C under argon. Prior to injection, the nanoliposomal IFPT plus Alum vaccine, hereafter called L-IFPTA+, was brought to RT and carefully mixed.
Animal and cell line
A total of 20 female C57BL/6 mice (4–6 weeks old) were bought from the Pasteur Institute of Tehran, Iran and fed with ad libitum access to purified water and a commercial stock diet. All mice were housed in a pathogen–free animal house at a temperature of 22 ±1°C with a 12 : 12-h light: dark cycle and maintained under a relative humidity of 50 ±10%. Animal care was performed in accordance with welfare guidelines established by the Institutional Ethical Committee and Research Advisory Committee of Mashhad University of Medical Sciences. At the end of the experiment all animals were euthanised by CO2 inhalation.
The B16F0 melanoma cell line was provided from the Pasteur Institute of Tehran, Iran and cultured in RPMI-1640 medium containing 10% FBS and supplemented with penicillin (100 IU/ml)/streptomycin (100 mg/ml). The cells were incubated at 37°C with a 5% CO2/95% air humidified atmosphere.
Vaccination program
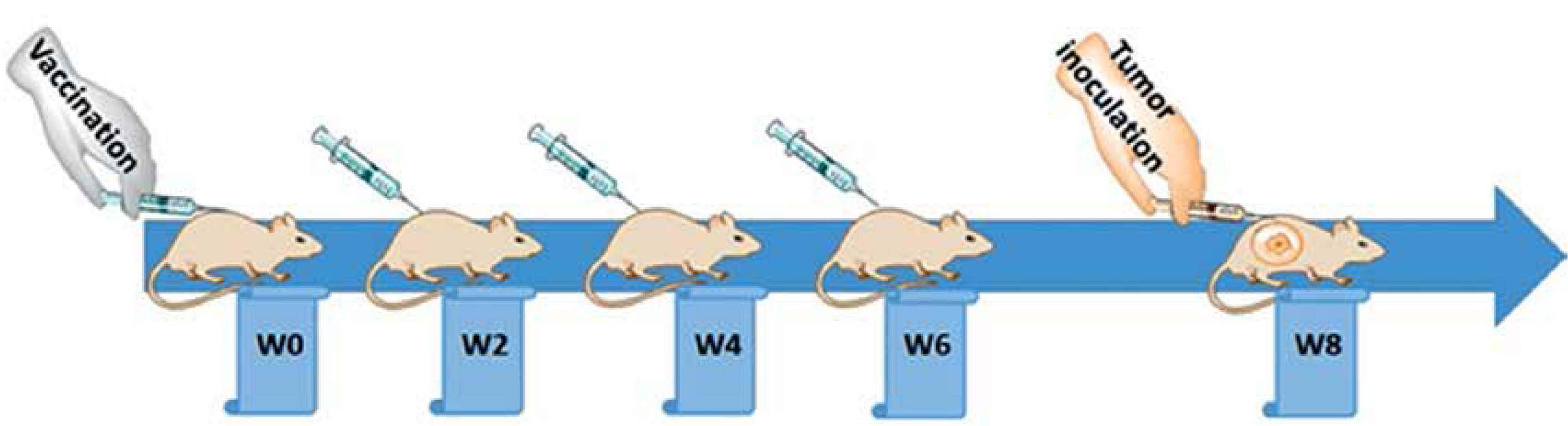
Following one week of taming prior to the experimental procedures, the mice were randomly arranged into two groups: a vaccine group (n = 10) and an untreated group (n = 10). The vaccination was primed at week 0 (W0) and followed by three boosters (W2, W4, and W6), in a bi-weekly interval via subcutaneous administration (Figure 2), while untreated mice simultaneously received phosphate-buffered saline (PBS). The tail vein bleeding was performed 2 weeks after each vaccination for the titration of plasma anti-PCSK9 antibody.
ELISA measuring of plasma anti-PCSK9 peptide antibody
To determine the titre of anti-PCSK9 antibodies, plasma samples were collected and analysed by ELISA method. Briefly, PCSK9 peptide at the concentration of 5 μg/ml in 0.1 M NaHCO3 (pH 9.2–9.4) was coated overnight in a 96-well Nunc-MaxiSorp plate. Free binding sites were then blocked by the incubation with blocking buffer (1× PBS, 1% BSA) for 1 h at 37°C. Diluted plasma (1 : 400 in 1× PBS/0.1% BSA/0.05% Tween-20) was added, serially diluted 1 : 4, and incubated for 1 h at 37°C. Each ELISA plate contained a standard antibody as an internal control. For the detection, biotinylated anti-mouse IgG (H + L) (Sigma-Aldrich; 1 : 1000) in 1× PBS/0.1 % BSA/0.1% Tween-20 was applied and incubated for 1 h at 37°C. As a next step, horseradish peroxidase coupled to streptavidin (Roche) was added (30 min, 37°C) followed by the addition of the substrate 2,2′-Azinobis [3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt (ABTS) (Sigma-Aldrich) (15 min, RT). The optical density (OD) at 450 nm was measured with a microwell plate reader (Sunrise, Tecan, Switzerland), and the titres were defined as the dilution factor referring to 50% of the maximal optical density (ODmax/2). The mean titres ± SD of all animals per group are presented.
Plasma PCSK9 quantification
Plasma PCSK9 concentration in the vaccinated mice was measured by CircuLex rat PCSK9 ELISA (CircuLexTM, Cy-8078, MBL, Woburn, MA) according to the manufacturer’s instructions. Briefly, 100 μl of the diluted 1 : 100 plasma samples was added on a 96-well microplate and incubated for 1 h at RT. A HRP-conjugated anti-PCSK9 antibody was added for 1 h followed by the substrate reagent and stop solution, all at RT. Optical density was detected at 450 nm with a Microwell plate reader (Sunrise, Tecan, Switzerland). A standard curve provided by the supplier was defined to measure PCSK9 concentration.
PCSK9 inhibition analysis
To evaluate inhibition of mice PCSK9 by vaccine-induced antibodies, interaction of generated-antibodies with PCSK9 was assayed. For this purpose, the same kit CircuLex rat PCSK9 ELISA was used, but in turn of HRP-conjugated anti-PCSK9 antibody, detection was performed with HRP-conjugated anti-mouse IgG (H+L) (Sigma Aldrich; dilution 1 : 5000) incubated for 1 h at RT, followed by the substrate reagent and stop solution provided by the supplier. The OD was detected at 450 nm with the Microwell plate reader.
In vitro evaluation of inhibiting PCSK9-LDLR interaction
CircuLex PCSK9-LDLR in vitro binding assay kit (CircuLexTM, Cy-8150, MBL, Woburn, MA) was employed to analyse the ability of vaccine-generated antibodies for inhibition of the PCSK9-LDLR interaction in vitro. Briefly, 100 μl of vehicle control or the plasma samples of vaccinated mice was added to a 96-well microplate pre-coated with a recombinant LDLR-AB domain, which contained binding site for PCSK9. Immediately after that, the reaction was initiated by adding a “His-tagged PCSK9 wild type” solution incubated for 2 h followed by adding a biotinylated anti-His-tag monoclonal antibody for 1 h at RT. Then, HRP-conjugated streptavidin was coated for 1-h at RT followed by the substrate reagent and stop solution. In this method, the higher amount of PCSK9-LDLR interaction is associated with higher ELISA OD, in which, in the presence of anti-PCSK9 antibody, this interaction is inhibited and consequently ELISA OD is decreased. A dose-response curve with appropriate serial dilutions of “His-tagged PCSK9 wild type” solution was drawn to measure accurate inhibition percentage of test samples.
Evaluation of in vivo anti-tumour efficacy
Two weeks after the last booster, the vaccinated and unvaccinated C57BL/6 mice were subcutaneously inoculated with B16F0 melanoma cells (5 × 105/50 μl PBS/mouse) into the right flank at day zero. Tumour growth was monitored in a three-day interval by calculating the tumour volume after measuring three orthogonal diameters with callipers according to the formula: Tumour volume (mm3) = (length × height × width) × 0.52. After the tumour mass was palpable (approximately 10 mm3) on day 10, the mice were randomly divided into four groups (5 mice/group) and subjected to different treatment protocols: (1) PBS (untreated control) group, which comprised unvaccinated tumour-bearing mice receiving single tail vein injection of saline buffer; (2) vaccine group, which comprised vaccinated tumour-bearing mice receiving a single tail vein injection of saline buffer; (3) the combination group which comprised vaccinated tumour-bearing mice receiving a single tail vein injection of Doxil (15 mg/kg); and (4) the Doxil (positive control) group, which comprised unvaccinated tumour-bearing mice receiving a single tail vein injection of Doxil (15 mg/kg).
To evaluate therapeutic efficacy, mouse body weight, tumour size, and survival were monitored every 3 days for 38 days. Animals euthanasia (CO2 inhalation) occurred on those with 4T1 tumour due to the following ethical considerations: body weight loss > 20% of initial mass, tumour volume greater than 2.0 cm in one dimension, or if the mice became sick and unable ambulate to reach food/water [47, 48]. For each mouse, the time to reach tumour volume above 1000 mm3 or the time to reach end point (TTE) as a response variable was calculated from the equation of the line obtained by exponential regression of the tumour growth curve. For each group, the percent of tumour growth delay (%TGD) was determined by calculating the difference between the average TTE of treatment group (T) and the average TTE of the control group (C), (%TGD = [(T – C)/C] × 100) [49]. For each treatment group, the percent of increased life span (%ILS) was measured based on the following formula: [(the average survival time of treatment group/the average survival time of control group × 100) – 100] [50].
Statistical analysis
Statistical analysis was performed using SPSS Statistics version 20 and GraphPad Prism version 7.04 software. Survival data were analysed using the log-rank (Mantel–Cox) test. Other comparisons were done using one-way ANOVA and Tukey post-hoc multiple comparison test. Values were expressed as mean ± SD or median for normally and non-normally distributed data, respectively. Results with p < 0.05 were considered as statistically significant.
Results
Characterisation of nanoliposomes
Physical properties of the free and IFPT-linked nanoliposomes are shown in Table II. The free and IFPT-linked nanoliposomes had a size range from 140 nm to 170 nm in diameter, with polydispersity index values of < 0.2, showing homogeneity of particles. Zeta potential measurement indicated negative charge on the surface of nanoparticles.
TLC and HPLC analysis of DSPE-PEG-IFPT micelles
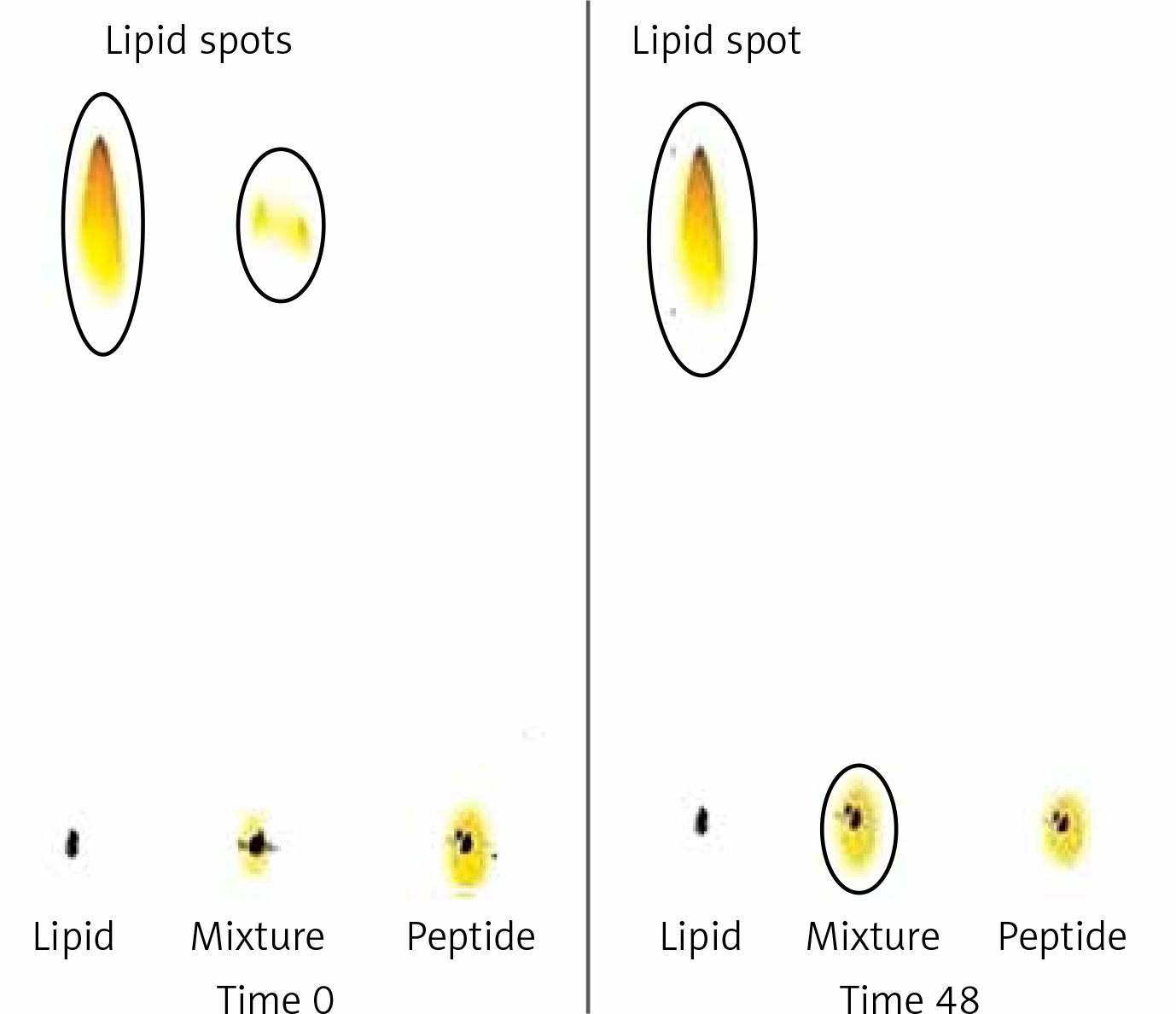
Conjugation of DSPE-PEG-Mal linker and the IFPT peptide was confirmed by evaluating the extinction of DSPE-PEG-Mal spot (lipid spot) on the TLC plate, following a 48-h reaction (Figure 3). Afterward, DSPE-PEG-IFPT micelles were prepared, and the efficiency of conjugation was measured using HPLC analysis, indirectly, by quantification of the free peptide content of the micelle sample. HPLC results indicated the conjugation of 96% of the initial IFPT peptides to DSPE-PEG-Mal linker (Figure 4).
Figure 3
Assessment of conjugation between DSPE-PEG-Mal and the IFPT peptide at the time zero and 48 h after starting of reaction. Lipid (DSPEPEG- Mal) is dissolved in the mobile phase and ascends to the top of the TLC plate (spots in the top of the left and middle lines), but peptide is bound to the silica and remains in the spotting point (the middle line). After 48, lipid bound to peptide and stayed in the point of spotting, and therefore the lipid spot on the top of the reaction mixture line disappeared, indicating the conjugation of the IFPT peptide and DSPE-PEG-Mal linker
Induction of PCSK9 antibodies in C57BL/6 mice by L-IFPTA+ vaccine
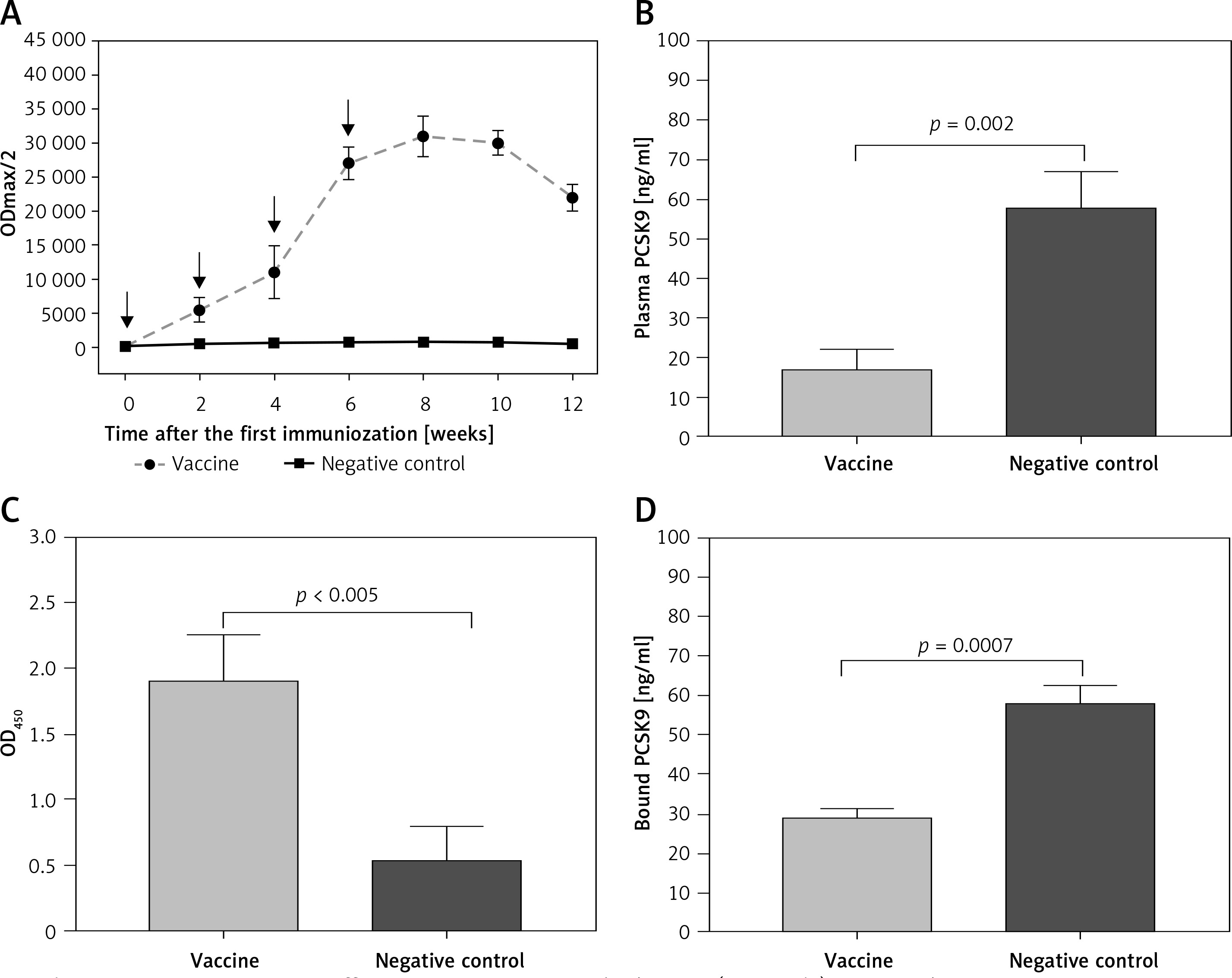
IFPT-exposing nanoliposomal vaccine was found to induce high titres IgG antibody against PCSK9 peptide in C57BL/6 mice upon four vaccinations in biweekly intervals. Further, long-term analysis revealed that anti-PCSK9 antibody titre peaked at week 8 and remained constant up to week 10, and then exhibited a diminishing trend (Figure 5 A).
Figure 5
AntiPCSK9 vaccine efficacy. A – AntiPCSK9 antibody titres (ODmax/2) generated upon 4 immunisations in a bi-weekly interval (signed by arrows) have been evaluated upon 12 weeks post prime immunisation. B – Plasma levels of PCSK9 in the vaccine and control group were 17 ±5 ng/ml and 58 ±9 ng/ml, respectively. C – Direct detection of antibodies bound to plasma PCSK9 in plasma samples from vaccinated and control mice. Increased OD450 is indicative for vaccine-generated anti-PCSK9 antibodies, which directly target PCSK9. D – In vitro PCSK9/LDLR binding assay. Plasma sample of vaccine group could decrease PCSK9 binding to LDLR by 50%, when compared with plasma sample of control group. Values are expressed as means ± SD (n = 3 replicates of the pooled samples of 10 mice per group). Significance compared to control values was analysed by unpaired 2-tailed Student’s t-test
Specific targeting of plasma PCSK9 vaccine-induced PCSK9 antibodies
L-IFPT vaccine provoked antibodies that could directly and specifically target plasma PCSK9 in C57BL/6 mice. Plasma levels of PCSK9 in the vaccine and control groups were 17 ±5 ng/ml and 58 ±9 ng/ml, respectively (Figure 5 B). Compared with the control mice, plasma levels of PCSK9 was significantly decreased by 70% (–41 ±6 ng/ml, p = 0.009) in the vaccinated mice. To evaluate specific targeting, PCSK9 inhibition was evaluated using ELISA-based assay to detect vaccine-induced antibodies directly bound to plasma PCSK9. Therefore, plasma PCSK9 obtained from the vaccinated and control mice was captured onto murine antiPCSK9 antibodies-coated ELISA plates, and vaccine-induced PCSK9-bound murine antibodies were detected using an anti-mouse IgG antibody. As shown in Figure 5 C, the significantly higher OD450 signal indicated from plasma of vaccinated mice revealed a direct binding of vaccine-generated anti-PCSK9 antibodies to PCSK9.
PCSK9-LDLR binding inhibition by vaccine-induced PCSK9 antibodies
In vitro PCSK9-LDLR binding assay revealed that in the presence of plasma anti-PCSK9 antibodies, interaction between murine PCSK9 and LDLR was inhibited. Plasma obtained from the L-IFPTA+ group could significantly decrease PCSK9 binding to LDLR by 50%, compared with the plasma from the control group (Figure 5 D).
Efficacy study in mice bearing B16F0 melanoma tumours
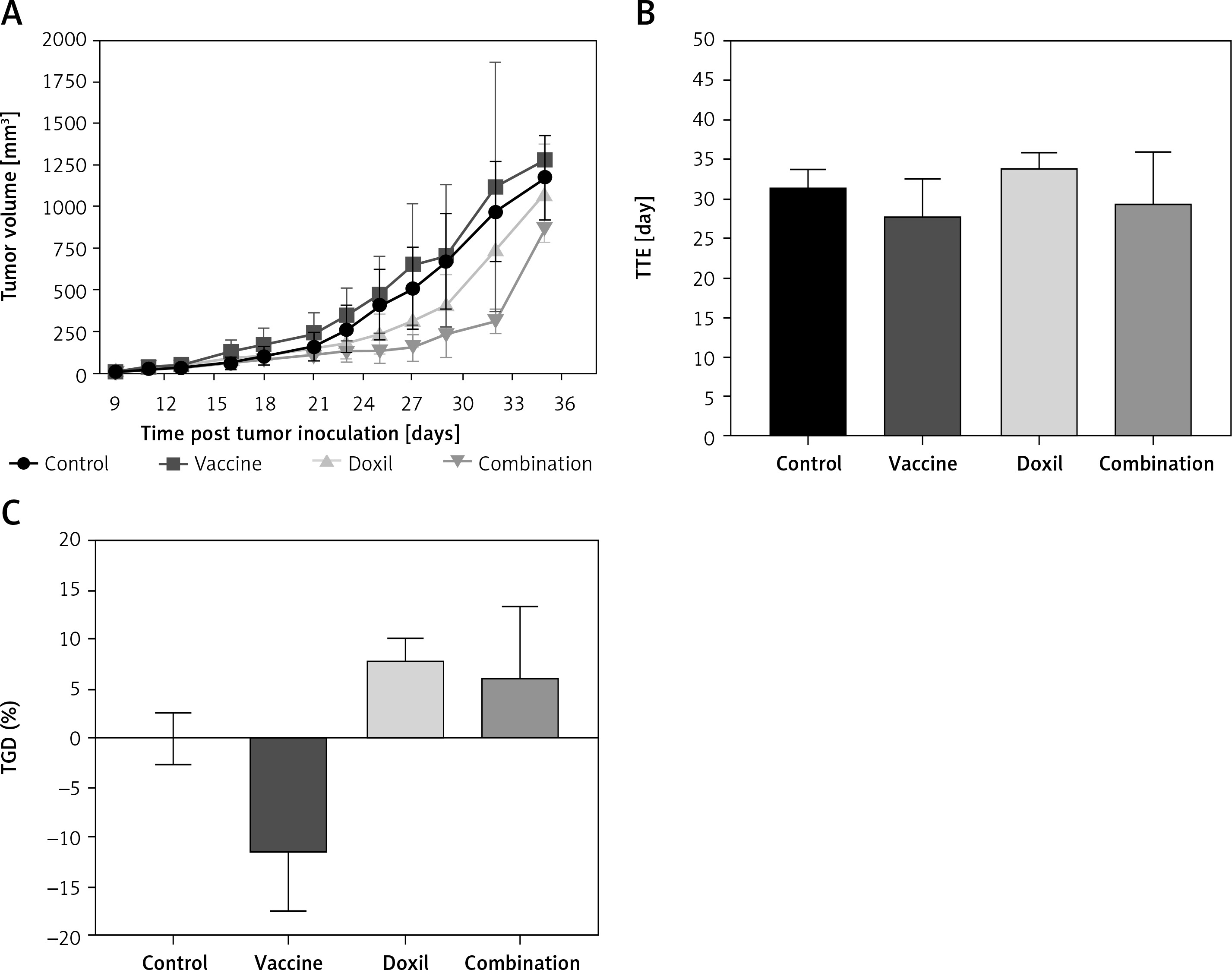
The effect of PSCK9 inhibition on skin cancer progression was evaluated using a model of B16F0 melanoma tumour developed in the vaccinated C57BL/6 mice. The protective effect of the liposomal-antiPCSK9 vaccine on tumour-bearing mice was studied by monitoring body weight alterations, tumour growth rate in terms of mean tumour size (mm3), and survival. The weight monitoring curve (Figure 6 A) and the integrated areas under the body weight curve (AUCbody weight) over 36 days (Figure 6 B) showed that body mass was not significantly different between the vaccine, liposomal doxorubicin (nanoliposomal doxorubicin), and control group. To determine the endpoint therapeutic potential of the liposomal-antiPCSK9 vaccine in tumour-bearing C57BL/6 mice, time to reach endpoint (TTE), and percentage of tumour growth delay (%TGD), as well as median survival time (MST) and increase life span (ILS) were measured (Table III). As revealed by TTE and %TGD, tumour growth was not significantly different between the vaccine, liposomal doxorubicin, and control groups (Figure 7 and Table III). TTE values of the vaccine, liposomal doxorubicin, and control groups were 28 ±5, 34 ±2, and 31 ±2 days, respectively. Percentage TGD values of the vaccine, Doxil, and control groups were 11.5 ±15.4%, 7.75 ±6.5%, and 0 ±7.5, respectively.
Table III
Therapeutic efficacy data of different treatments in C57BL/6 mice bearing B16F0 tumour
| Groups | TTEa (mean ± SD) [days] | TGDb (%) | MSTc [days] | ILSd (%) |
|---|---|---|---|---|
| Control | 31 ±2 | – | 32 | – |
| Vaccine | 28 ±5 | –11.5 | 32 | 0 |
| Doxil | 34 ±2 | 7.8 | 35 | 9.4 |
| Combination | 30 ±6 | –6.1 | 32 | 0 |
Figure 6
A – The weight monitoring curve exhibits point by point alterations of the body weight during 36 days in the control, vaccine, and Doxil group. B – The integrated areas under the body weight curve (AUCbody weight) over 36 days demonstrate overall weight changes. The body weight loss was not significantly different between the vaccine, Doxil, and the control group. Animal body weight was measured every 3 days. Data are presented as the mean ± SD (n = 8). P < 0.05 was considered as the level of statistical significance
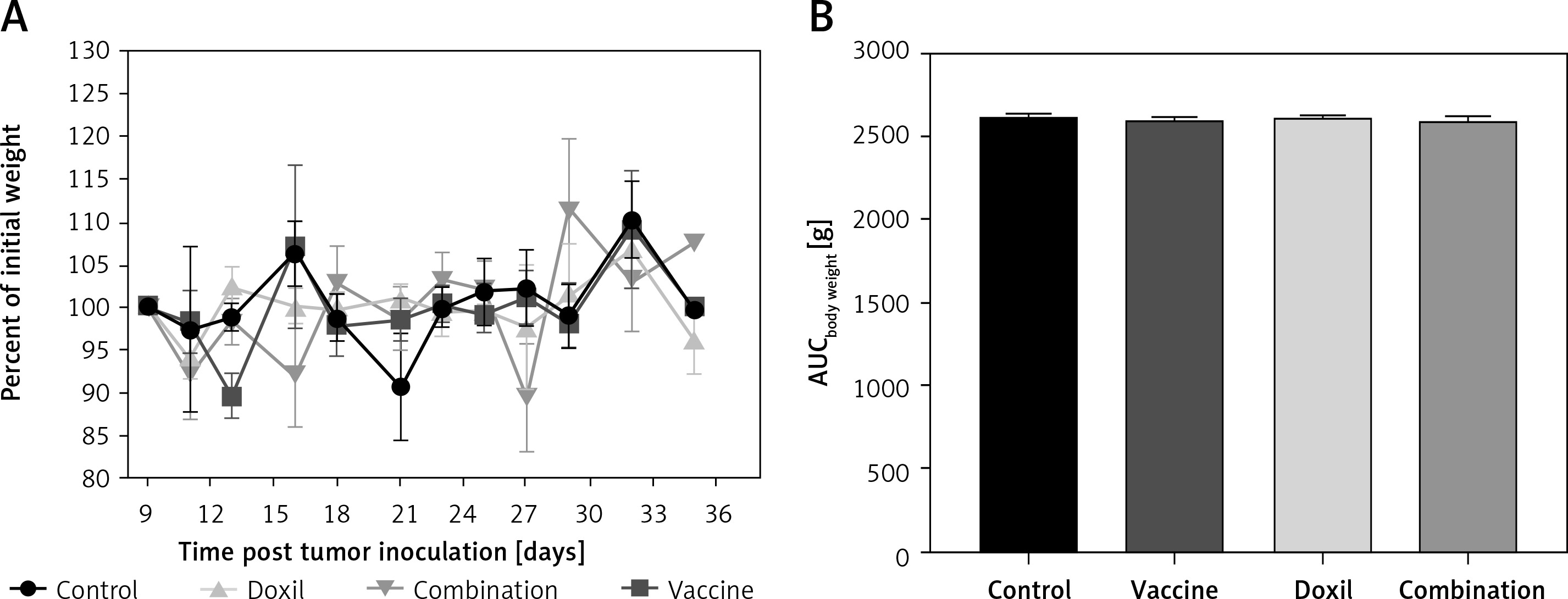
Figure 7
The tumour growth curve (A) shows increase of tumour size during 36 days in the control, vaccine, and Doxil group. Measuring the time to reach endpoint (TTE) (B) and tumour growth delay (TGD) (C) showed no significant difference between the studied groups. Tumour volume (mm3) was measured every 3 days. Data are presented as the mean ± SD (n = 8). Results with p < 0.05 were considered as statistically significant
Kaplan-Meier curves (Figure 8) showed that the survival of both the vaccine and liposomal doxorubicin treated groups was not significantly different from the control mice (p > 0.05, log-rank test). The MST of the vaccine, liposomal doxorubicin, and control group was 32, 35, and 32 days, respectively. Further analysis did not reveal any difference between the ILS indices of the vaccinated group and the other groups (Table III).
Figure 8
Kaplan-Meier curves exhibit the survival rate of the control, vaccine, and Doxil group. There was no significant difference between the compared groups. Data are presented as the mean ± SD (n = 8). P < 0.05 was considered as the level of statistical significance
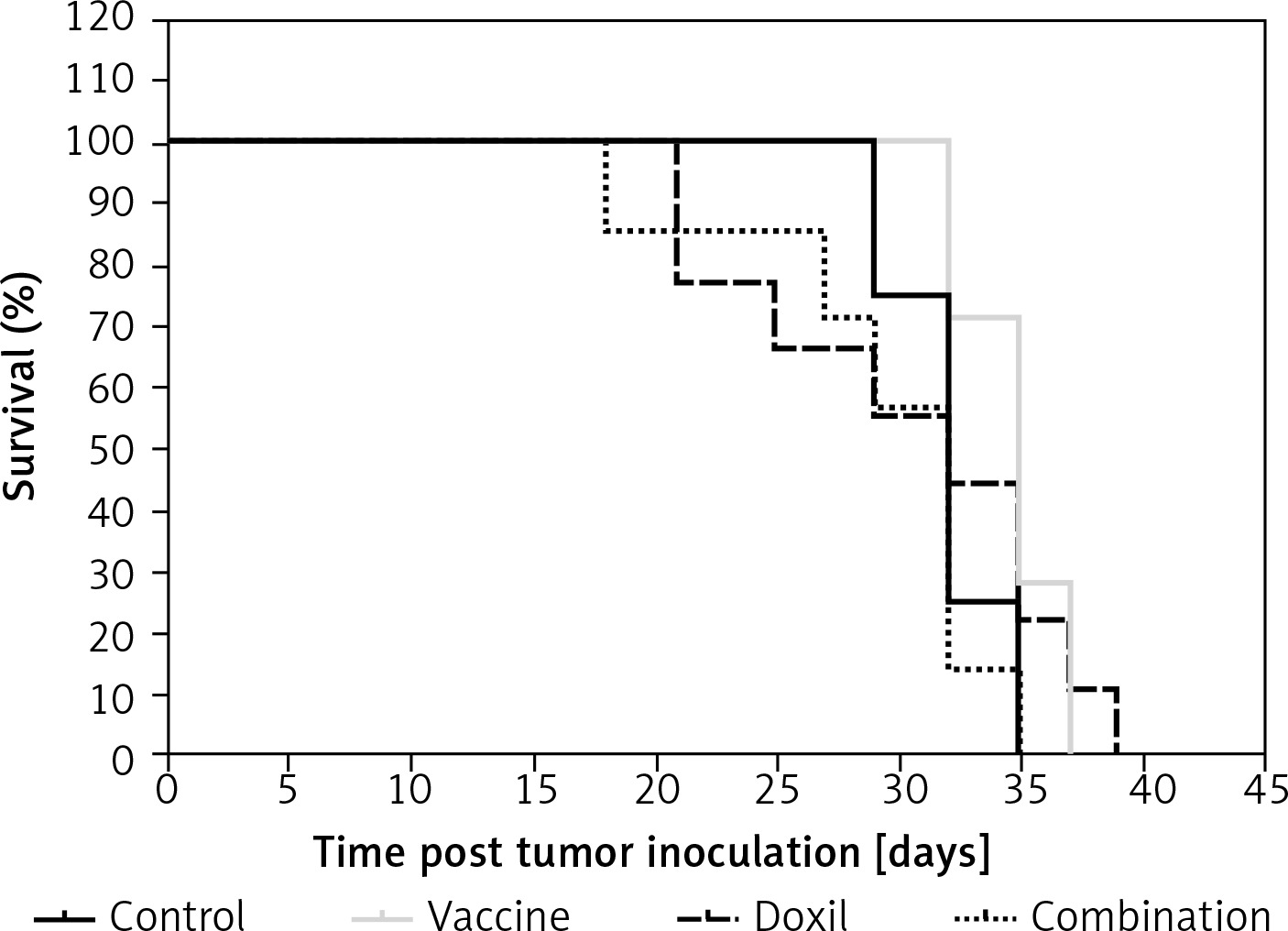
To evaluate the effect of PCSK9 inhibition on tumour chemotherapy, a vaccinated group were taken under single-dose Liposomal doxorubicin treatment (called the combination group) and compared with the group only vaccinated and with the group treated with liposomal doxorubicin alone. It was found that body weight change was not significantly different between the combination, vaccine, liposomal doxorubicin, and control groups (Figures 6 A and B). Values of TTE and TGD% revealed that the tumour volume in the combination group was not significantly different from the vaccine, liposomal doxorubicin, and control group (Figure 7 and Table III). Kaplan-Meier curve analysis using log-rank test showed that there was no significant difference in MST and ILS indices between the combination group and the other groups (Figure 8 and Table III).
Discussion
We recently reported that L-IFPTA+ vaccine can effectively inhibit PCSK9 function through suppression of PCSK9/LDLR interaction via induction of antiPCSK9 antibodies in vaccinated BALB/c mice [41]. Although disrupted cholesterol homeostasis is known to play an important role in melanoma and other cancer types [1–10], the effects of lipid lowering agents, such as PCSK9 inhibitors, on cancer progression are unclear. Our previous experimental studies showed that L-IFPTA+ vaccine-mediated PCSK9 inhibition not only caused no harmful effects but also could moderately suppress tumour growth and improve lifespan and survival in mice bearing breast [51] and colon cancers [52]. Importantly, it has been found that cholesterol can exert differing effects dependent on cancer type; therefore, evaluating the effect of PCSK9 inhibition on other cancer types [53], e.g. melanoma, is necessary.
The findings of the present study revealed that L-IFPTA+ vaccine could provoke functional anti-PCSK9 antibodies (Figure 5 A) that reduce plasma level and activity of PCSK9 (Figures 5 B and D) through specific and direct targeting of PCSK9 (Figure 5 C) in C57BL/6 mice. However, the L-IFPTA+ vaccine could not inhibit tumour growth (Figure 7), nor did it prolong survival (Figure 8) in melanoma tumour-bearing mice. When compared with the liposomal doxorubicin and control groups, PCSK9 vaccination did not change TTE, TGD%, MST, or ILS, suggesting that L-IFPTA+ vaccine did not exacerbate tumour behaviour and lifespan of mice bearing melanoma.
Although there is no evidence on the effects of PCSK9 inhibitors in cancerous conditions, recent studies have shown that PCSK9 variants with LDL-lowering mutations mimicking PCSK9 inhibitors are associated with a lower risk and occurrence of cancer [36, 54]. Such inconsistency between results of our study and others is probably due to different modalities of PCSK9 inhibition. We inhibited circulating PCSK9, in which the LDLR-dependent effect of PCSK9 was affected, while genetic mutations possessing LDL-lowering variants can modulate intracellular activity of PCSK9 beyond its regulatory effect on LDLR, such as regulation of cell cycle and apoptosis [55, 56]. Additionally, L-IFPTA+ vaccine, when combined with liposomal doxorubicin chemotherapy, did not aggravate the effects of chemotherapy nor improved the anti-tumour effect of liposomal doxorubicin. There was also no significant difference in lifespan and MST of mice in the vaccine and combination groups.
In conclusion, although L-IFPTA+ vaccine-mediated PCSK9 inhibition could not decrease the growth of melanoma tumours and improve the survival of tumour-bearing animals, it did not aggravate tumour behaviour in melanoma-bearing mice either alone or when combined with chemotherapy (liposomal doxorubicin). Importantly, further preclinical and clinical investigations are needed to confirm the efficacy and safety of therapeutic inhibition of PCSK9 in patients with different types of cancer.