Introduction
Neonatal hypoxic-ischemic brain damage (HIBD) results from inadequate oxygen and blood supply around the time of birth and can lead to severe morbidity and mortality [1]. Up to 25–60% of survivors still have early mortality or severe disabilities, such as mental retardation, cerebral palsy, epilepsy or learning disabilities [2]. While mild hypothermia is currently the standard treatment for perinatal HIBD, its efficacy varies, and many infants do not respond adequately, underscoring the need for alternative treatments [3, 4]. However, the clinical trials show that full-term infants affected by mild hypoxic-ischemic encephalopathy show the best response to hypothermia therapy, while newborns affected by moderate to severe encephalopathy do not seem to show high efficiency or no response at all, and about 55% of newborns receiving mild hypothermia therapy are still not effectively protected [5]. Therefore, there is an urgent need to develop more effective methods to treat HIBD.
A wealth of evidence from in vivo and in vitro studies has highlighted the neuroprotective properties of estradiol (E2), which include anti-apoptotic, anti-inflammatory, and anti-oxidative effects. Astrocytes, the most abundant glial cells in the central nervous system, provide crucial support to neurons and represent a potential target for therapeutic intervention following cerebral ischemia [6]. As early as 15 years ago, researchers proved that 17β estradiol can significantly reduce hypoxic-ischemic brain damage and reduce infarct area in neonatal rats [7], but its specific mechanism is still unclear.
Astrocytes are the most abundant cell types in the central nervous system [8]. Under normal physiological conditions and after ischemia, astrocytes support neurons by providing antioxidant protection, eliminating metabolic substrates and glutamic acid of neurons, and providing energy for neurons through lactic acid shuttle [9]. So, astrocytes play an important role in nutritional support for neurons [10, 11]. Although astrocytes are sometimes more elastic than neurons under hypoxic-ischemic conditions, even if astrocytes are not dead, anoxia and ischemia will lead to the impairment of astrocyte function, which will amplify the death of neurons [10, 12]. It has been found that improving the function of astrocytes after ischemic brain damage can improve the survival rate of neurons [10, 13]. Exploring the improvement of astrocyte function after cerebral ischemia and enhancing support for neurons may become a new target for treating ischemic brain damage.
G protein-coupled estrogen receptor 1 (GPER1), a specific estrogen receptor, plays a critical role in rapid estrogen signaling and has been implicated in the neuroprotective effects of E2 [14]. Numerous studies have explored its role in the neuroprotective mechanism of hormones. For example, the agonist G1 of GPER1 upregulates the expression of the GPER in the hippocampus of aged female rats, and counteracts the effects of aging on animal anxiety and depression [15]; GPER1 is also involved in the neuroprotective effect of E2 in local and global ischemic models, which is manifested by reducing the infarct volume and the release of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and IL-6 in ischemic penumbra [16]. In addition, GPER mediates the up-regulation of glutamate transporter GLT-1 and the uptake of glutamate by astrocytes [17], which may reduce excitotoxic nerve damage.
As one of the downstream signaling pathways of GPER1, the PI3K/AKT pathway, a key downstream signaling route of GPER1, is known to promote cell survival and inhibit apoptosis, with nuclear factor-κB (NF-κB) being a significant player in the inflammatory response and cell death following ischemic injury [18]. When cells are exposed to oxidative stress, the PI3K/AKT signaling pathway is activated, which will subsequently change the downstream signal cascade and activate the downstream signal factor NF-κB [19]. After being activated, NF-κB participates in regulating inflammation, apoptosis and ischemia-reperfusion damage [20]. Many inflammatory factors also can activate NF-κB in the inflammatory response, such as TNF-α, IL-1 and IL-6. It has been observed in many studies that E2 can play a neuroprotective role by activating the PI3K/Akt pathway. For example, it was found that E2 promotes the development of midbrain neurons through the PI3K/Akt pathway [21]. It has also been reported that E2 can activate the PI3K/Akt pathway in an adult rat global cerebral ischemia model, reduce apoptosis of hippocampal neurons and play a role in brain protection [22]. Yang et al. found that activation of GPER1 can protect neurons from excitotoxicity through NF-κB signaling [23].
E2, GPER1 and astrocytes play an important role in ischemic brain damage and other central nervous system injuries. However, the relationship among astrocytes, GPER1 and the AKT/NF-κB pathway during the neuroprotective effect of E2 on neonatal rats after HIBD is unclear. Understanding the interplay among E2, GPER1, astrocytes, and the AKT/NF-κB pathway may uncover new avenues for enhancing post-ischemic astrocyte function and present novel therapeutic approaches for HIBD.
Material and methods
Animal experiments
Protocol
The protocol for animal experimentation received approval from the Experimental Animal Committee of Linyi Maternal and Child Health Hospital (approval number: [2022]006). Sprague-Dawley (SD) neonatal rats were procured from Beijing Huafu Biotechnology Co., Ltd. (license number: SCXK (Beijing) 2019-0008) and were housed at the Experimental Animal Center of Linyi Maternal and Child Health Hospital (license number: SYXK [Lu] 2021-0007). The animals were reared under a 12-hour light-dark cycle, at a controlled ambient temperature of 25 ±2°C and a relative humidity of 60–80%. Reverse osmosis (RO) purified water was provided ad libitum.
Experimental grouping and drug intervention
Neonatal SD rats were randomly assigned to one of eight groups: Sham + vehicle, Sham + E2, Sham + G15, Sham + E2 + G15, HIBD + vehicle, HIBD + E2, HIBD + G15, and HIBD + E2 + G15. On the second postpartum day, G15 was administered intraperitoneally at a dose of 1.46 mg/kg (stored at –80°C, with a DMSO concentration under 10%, and diluted with PEG300 + TW80 + 0.9%NS, with the final injection volume being less than 0.1 ml). E2 was injected intraperitoneally from the fifth postpartum day until 24 h after surgery at a dose of 0.1 mg/kg (E2 dissolved in DMSO and stored at –80°C). Vehicle groups received a mixture of 10% DMSO + 40% PEG300 + 5% TW80 + 45% NS as a control.
HIBD model construction
Corresponding to human full-term neonatal brain development, 7-day-old neonatal SD rats were utilized. The HIBD model was generated using the modified Rice-Vannucci method [24, 25]. This entailed double ligation of the left common carotid artery (CCA). The sham-operated group had the CCA exposed without ligation or hypoxic treatment. Following anesthesia, the rat’s neck was disinfected, and a midline incision allowed for CCA ligation. Post-operative rats were returned to their dams for a 1-hour recovery before being subjected to a hypoxic environment (8% O2 and 92% N2, flow rate of 1.5 l/min, at 37°C and over 75% humidity) for 4 h. Subsequently, rats were returned to their dams for continued care.
Neurobehavioral assessment
On the third post-hypoxia day, the neurobehavioral function of the neonatal rats was assessed in a tranquil environment employing established methods, including the righting reflex, negative geotaxis, and grip strength tests. Each test was performed three times, with the mean score being recorded.
TTC staining
Brain infarct volume was evaluated using 2,3,5-triphenyltetrazolium chloride (TTC) staining 72 h after HIBD. Anesthetized rats were decapitated, and the brains were rapidly frozen at –80°C for 3 min, then coronally sectioned and incubated in 1% TTC at 37°C for 15 min in the dark. Viable brain tissue appeared red, whereas infarcted areas remained pale. Infarct volume was calculated using the formula: [(volume of the contralateral hemisphere – volume of non-infarcted ipsilateral hemisphere)/volume of the contralateral hemisphere] × 100% [26].
Tissue protein extraction and Western blot detection
Brain tissues were collected 72 h after HIBD under deep anesthesia, homogenized in RIPA buffer, and centrifuged to obtain the supernatant. Protein concentration was determined using the BCA assay. For Western blot analysis, proteins were separated by SDS-PAGE, transferred onto PVDF membranes, and probed with primary antibodies against GPER1 (1 : 1000, GeneTex, USA, cat# GTX107748), Phospho-Akt (1 : 1000, CST, USA, cat#4060S), NF-kappaBp65 (1 : 1000, CST, USA, cat#8242S), cleaved-caspase3 (1 : 1000, Affinity, China, cat#AF7022), caspase-3 (1 : 1000, Abcam, UK, cat #13847), goat anti-rabbit IgG antibody (1 : 8000, genetex, USA, cat# gtx213110-01), β-actin (1 : 4000, Beyotime, China, cat# AF5003), Hinston H3 (1 : 2000, Proteintech, USA, cat#17168) followed by appropriate secondary antibodies. Chemiluminescent detection was used for visualization (Beyotime, China, cat# P0027).
Preparation of tissue sections
Brain tissues were perfused and fixed with 4% paraformaldehyde after HIBD, embedded, and sectioned. Sections were processed for histological analysis.
TUNEL staining of tissues
A TUNEL cell apoptosis detection kit (chromogenic method) (Beyotime, China, cat# C1098) was used to detect apoptotic cells in the ischemic penumbra area. The brain tissue sections were placed in a drying oven (70°C) to melt wax for 30 min in advance. After dewaxing and hydration, follow-up operations were according to the instructions of the kit. Finally, after DAB staining, the cell nucleus was stained with hematoxylin, the sections were sequentially dehydrated with alcohol gradient and rendered transparent with xylene before sealing and observation.
Cell experiment
Culture, purification and identification of astrocytes
Astrocytes were isolated and purified according to previous research [27]. When the cells were transferred to the third generation, they were inoculated on a 24-well culture plate for GFAP (Biyuntian, China, Cat#AG259) immunofluorescence staining to identify the purity of the cells.
Experimental grouping and drug intervention
Before drug intervention, the cells were divided into the following groups: Sham + vehicle, Sham + E2, Sham + G15, Sham + E2 + G15, OGD + vehicle, OGD + E2, OGD + G15, OGD + E2 + G15. According to the grouping, G15 intervention was applied 48 h in advance until OGD ended. E2 intervention was applied 24 h in advance until the end of OGD; the concentration of E2 working solution was 10 nM (dissolved in DMSO and stored in a refrigerator at –80°C; the concentration of DMSO working solution was less than 0.1%). Sham + vehicle and HIBD + vehicle groups used DMSO with the same concentration as E2 as a negative control.
Oxygen glucose deprivation/reperfusion (OGD/R)
After drug intervention, astrocytes were exposed to an anoxic and glucose-deficient environment to simulate anoxic and ischemic conditions. In short, the basal medium was replaced by glucose-free medium, then the cells were put in three-gas culture for 12 h (1% oxygen + 99% nitrogen). After that, they were replaced by normal complete medium and kept in normal culture conditions for 24 h.
CCK-8
CCK-8 was used to detect cell viability. The relative expression of TNF-α and IL-1β in cell supernatant was detected by enzyme-linked immunosorbent assay (ELISA).
Cells were inoculated into 96-well plates, with about 104 cells in each well. CCK-8 (Biyuntian, China, cat#C0041) was used to detect cell viability 24 h after OGD-R. TNF-α (Elabscience, China, cat#E-EL-R2856c) and IL-1β (Elabscience, China, cat#E-EL-R0012c) in the cell supernatant of each group were detected according to the operating instructions of the ELISA kit.
TUNEL for detecting apoptotic cells
Cells were inoculated into a 24-well plate containing cell slides. After 24 h of cell culture, drug intervention was applied based on cell grouping. After 24 h of OGD-R, the apoptosis of cells in each group was detected according to the instructions of the TUNEL kit (crondabio, China, cat#KCD-T1006). In short, 4% paraformaldehyde fixed cell slides for 30 min, 1% Triton X-100 membrane rupture was conducted for 5 min, then DNaseI reaction solution, TdT enzyme reaction solution and streptavidin-TRITC labeling solution were added dropwise, and finally DAPI staining solution was used to stain the nucleus and seal the film for observation.
The expression of p-AKT and NF-κBp65 was detected by immunofluorescence
Cells were inoculated into a 24-well plate containing cell slides, 24 h after OGD-R, fixed with 4% paraformaldehyde at 4°C for 30 min, exposed to 0.1% Triton X-100 at room temperature for 20 min, blocked with 5% BSA for 2 h, incubated with primary antibody (1 : 400, CST, USA, cat# 4060s) at 4°C overnight, fluorescent secondary antibody (1 : 200 bioshap China cat # BLO33A) was incubated at room temperature in the dark for 1 h, stained with DAPI for 10 min, and finally observed and photographed.
Statistical analysis
All experiments were repeated three times or more, and the data were expressed as mean and standard deviation. The experimental data were statistically analyzed by GraphPad Prism software (version 9.5, USA). The statistical significance was evaluated by one-way ANOVA. If the p-value was less than 0.05, the difference was considered significant.
Results
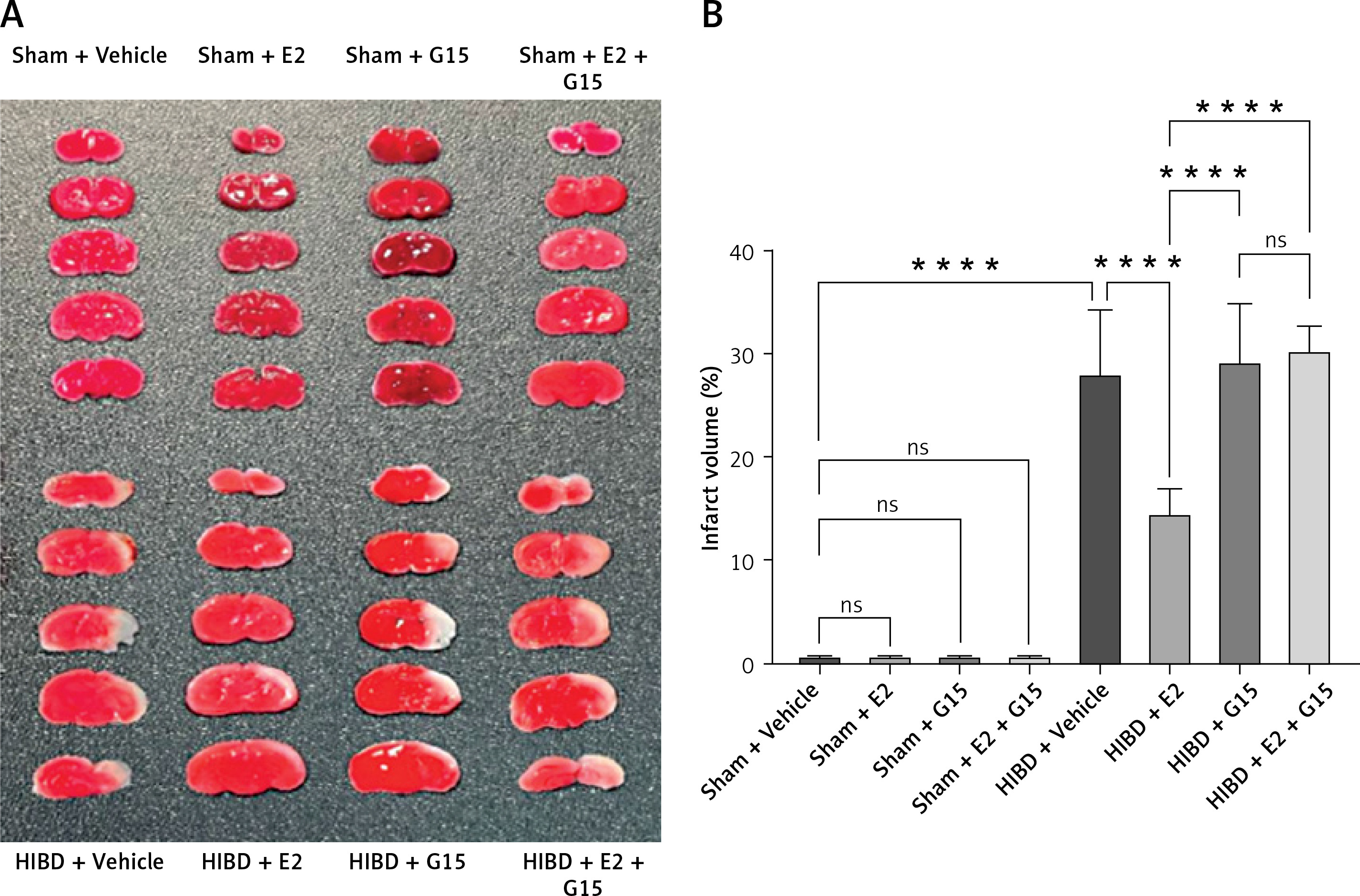
Estradiol (E2) intervention significantly mitigated cerebral infarction volume and behavioral impairments
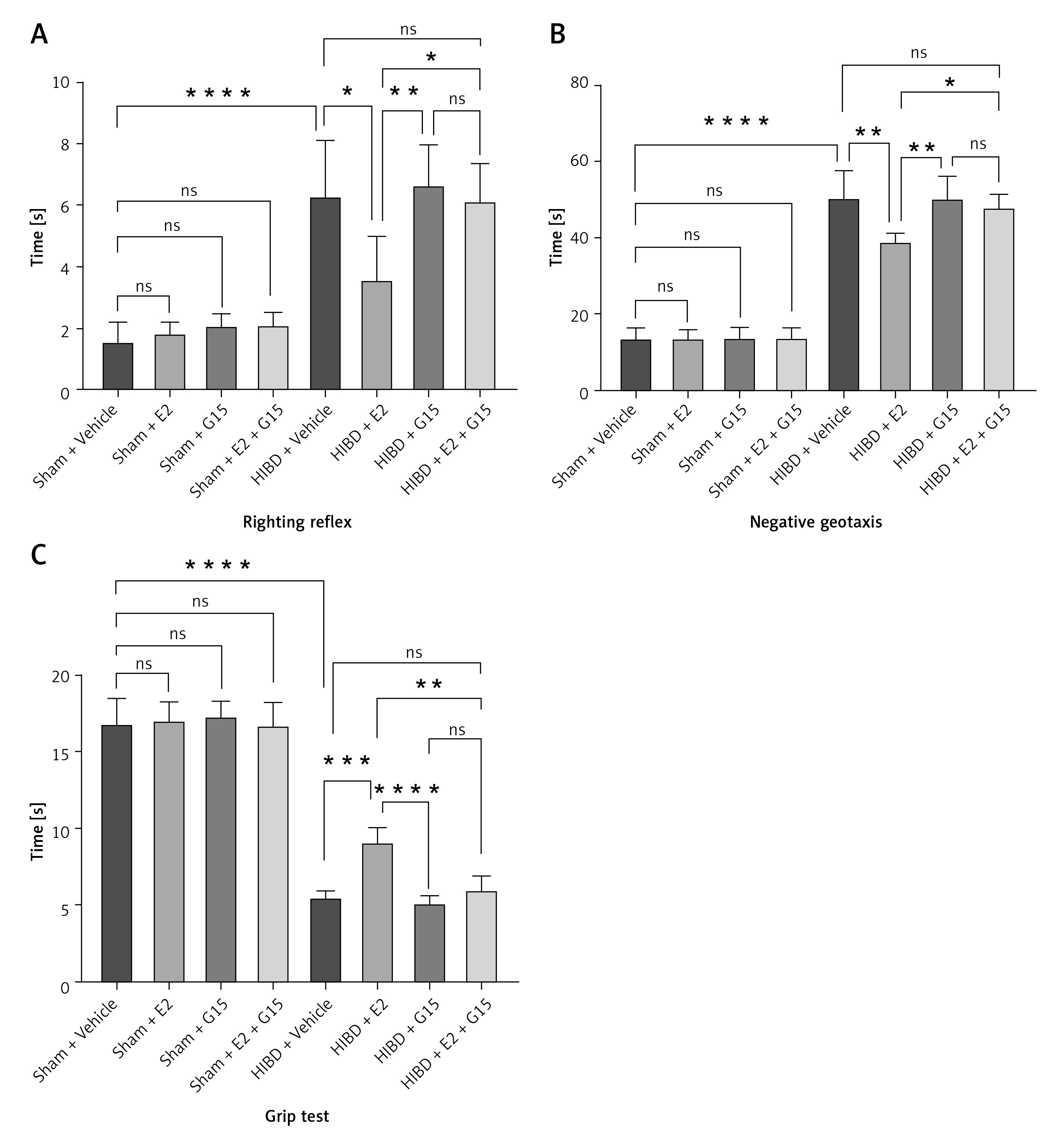
E2 administration markedly reduced the cerebral infarction volume in neonatal rats with HIBD, as indicated by TTC staining. Figure 1 illustrates that the infarction volume percentage in the HIBD + E2 group was significantly less than that in the HIBD + vehicle group. Furthermore, neurological function assessments using the righting reflex, negative geotaxis, and grip strength tests demonstrated that E2 ameliorated neurological impairments in HIBD-affected neonatal rats on the third post-surgery day (Figure 2). GPER1 inhibitor G15 reversed the neuroprotective effects of E2 neurobehavioral tests, including the righting reflex, negative geotaxis, and grip strength, indicated that G15 treatment negated E2’s neuroprotective benefits (Figures 2 A–C).
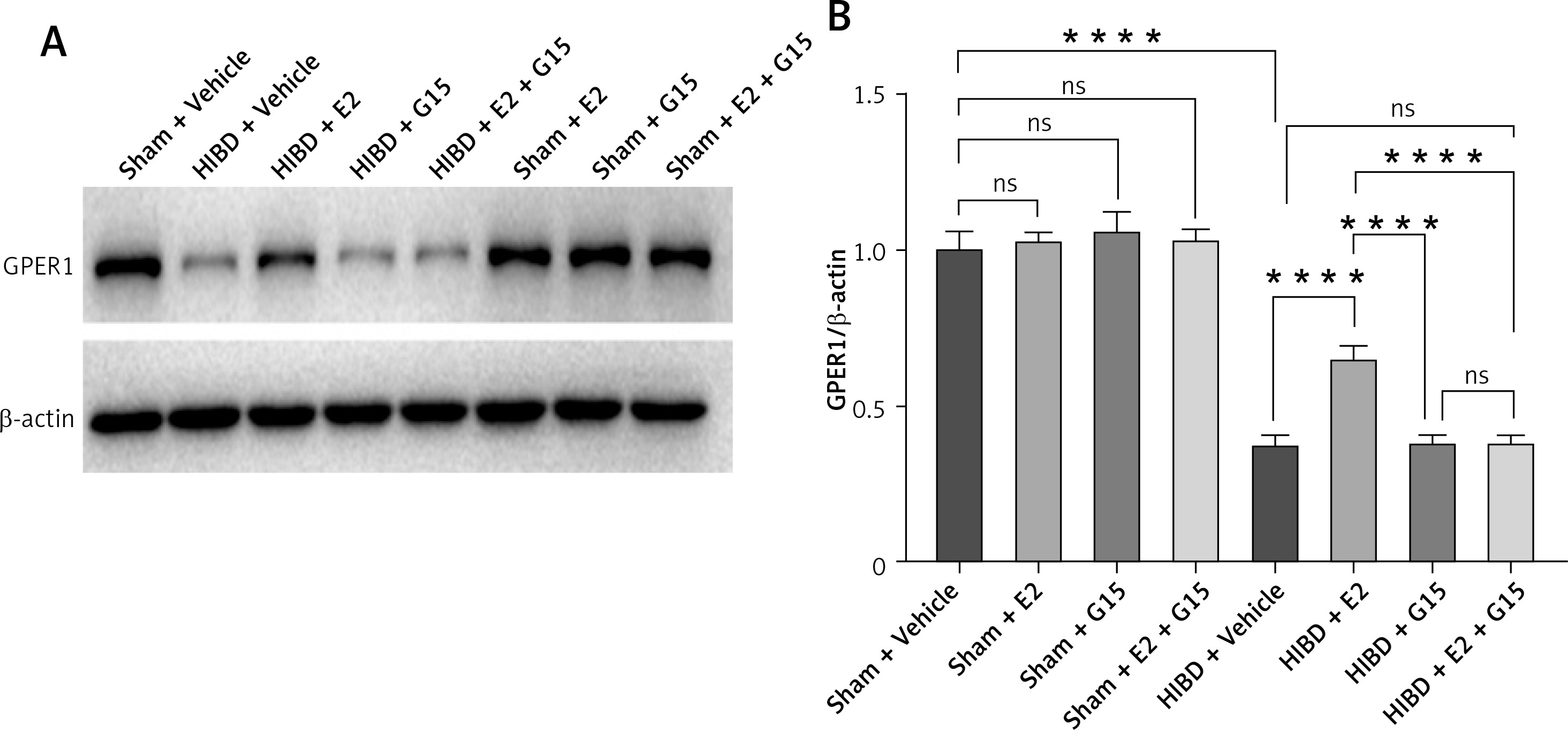
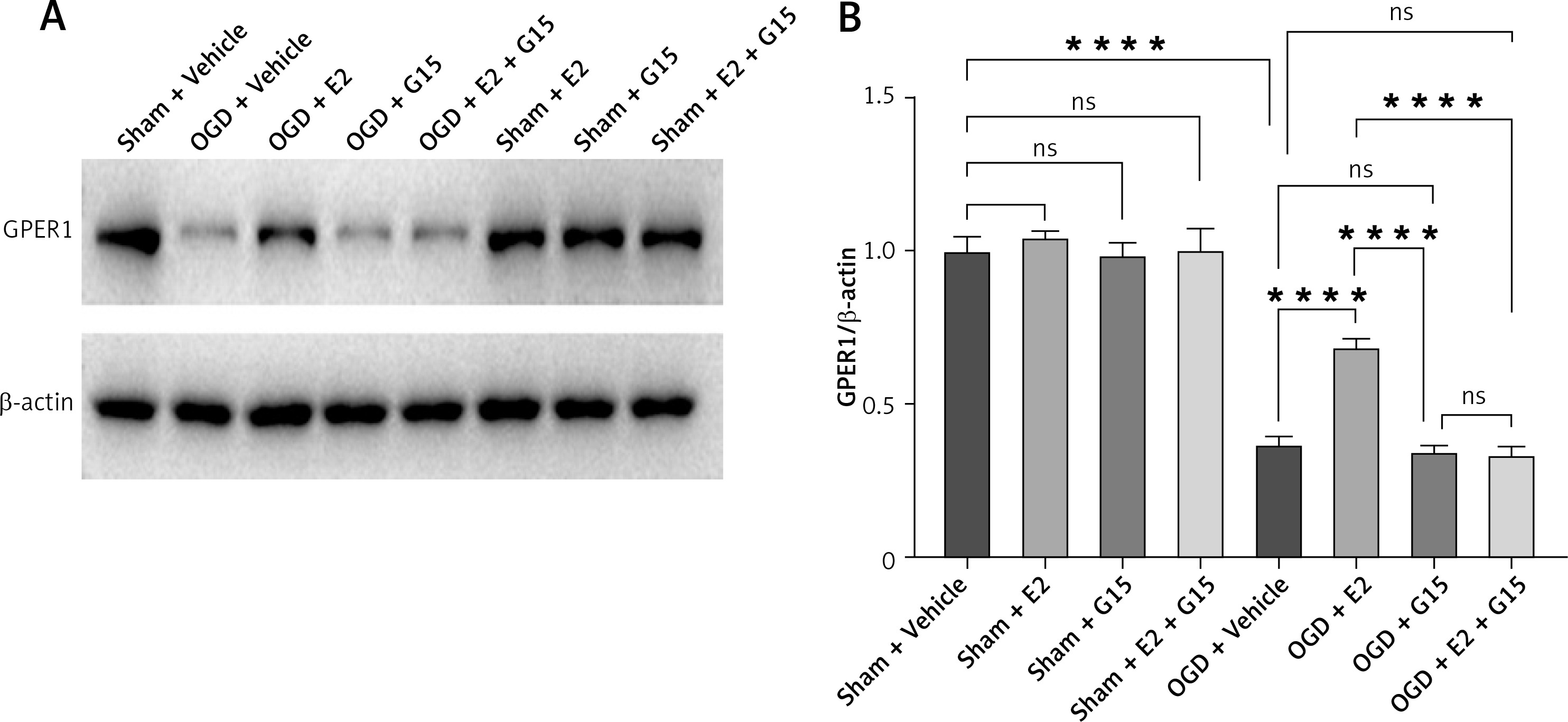
E2 increased GPER1 expression in HIBD models
HIBD induced a decrease of GPER1 expression, when E2 increased its expression. This effect was partly blocked by G15 (Figure 3).
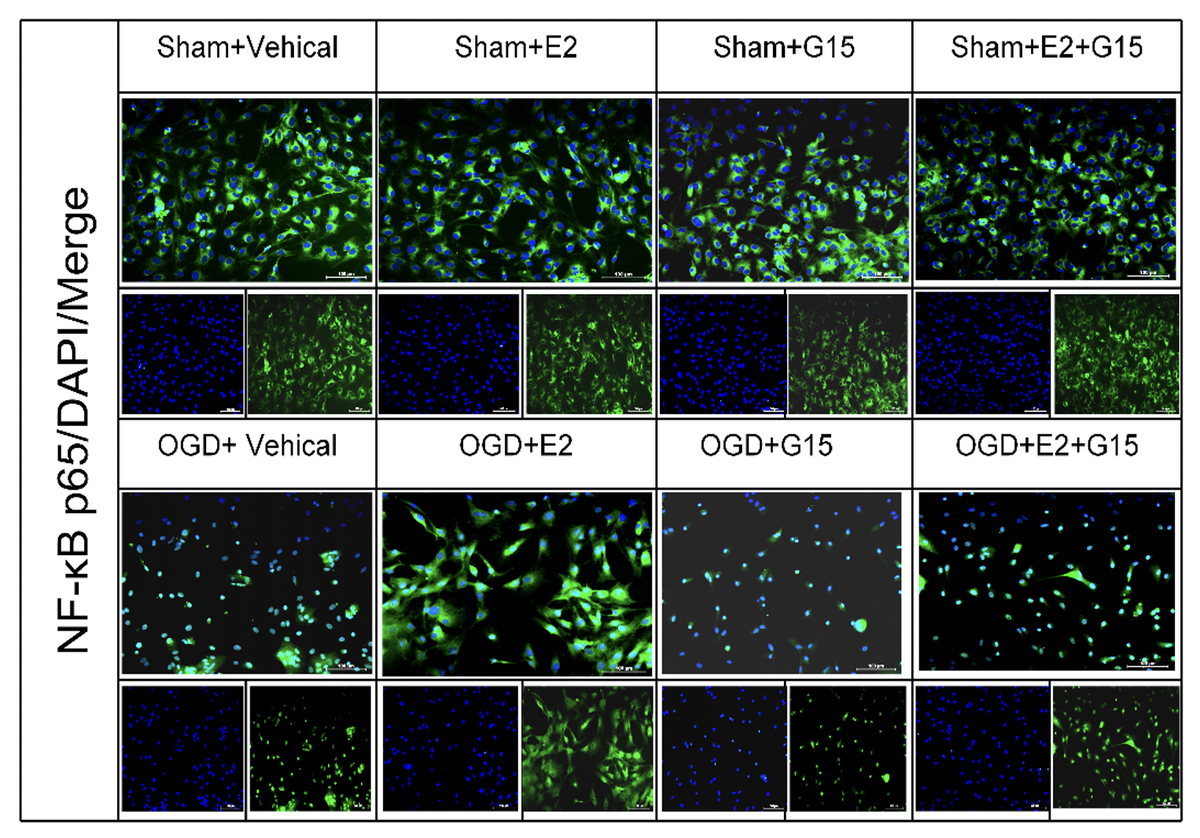
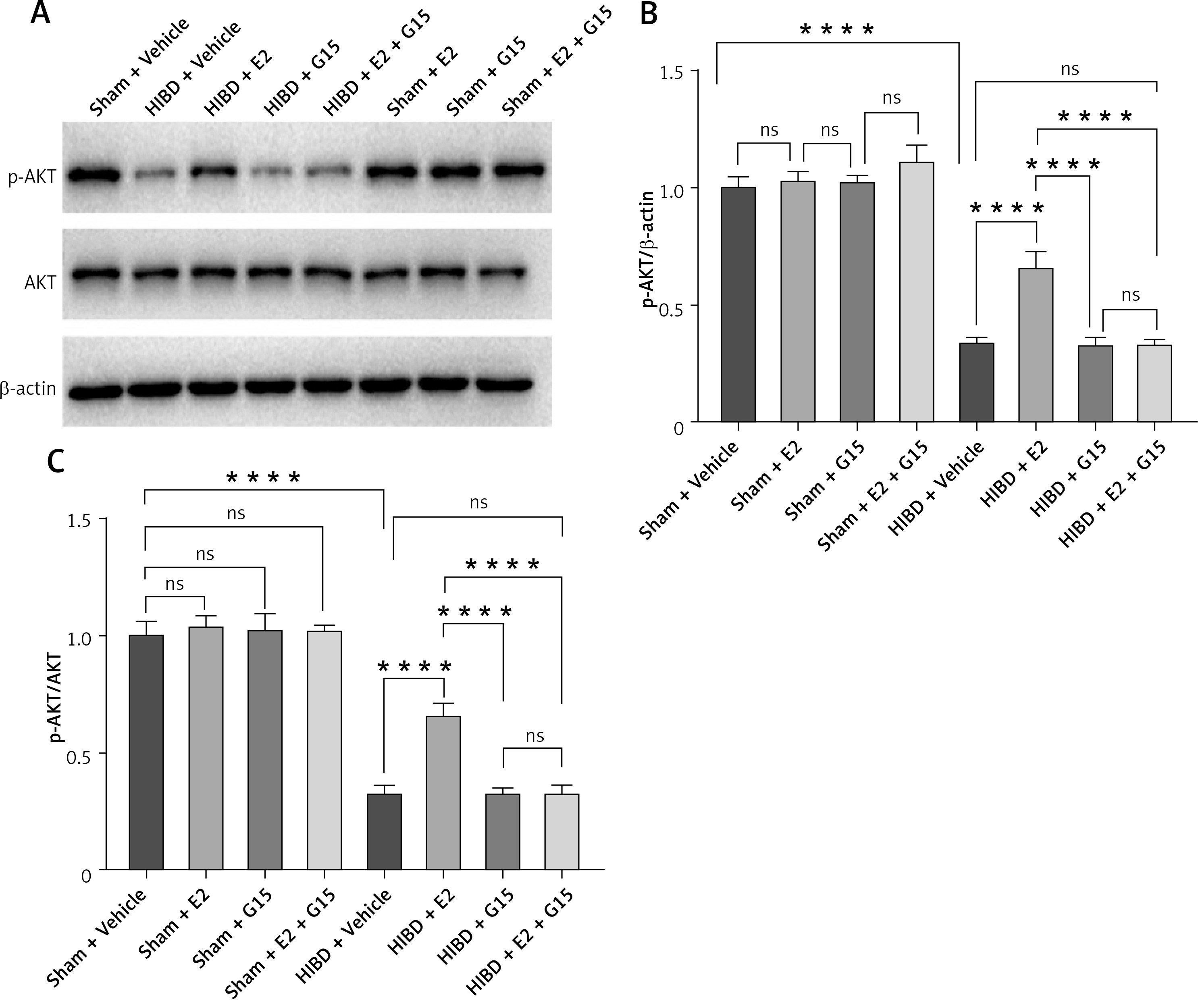
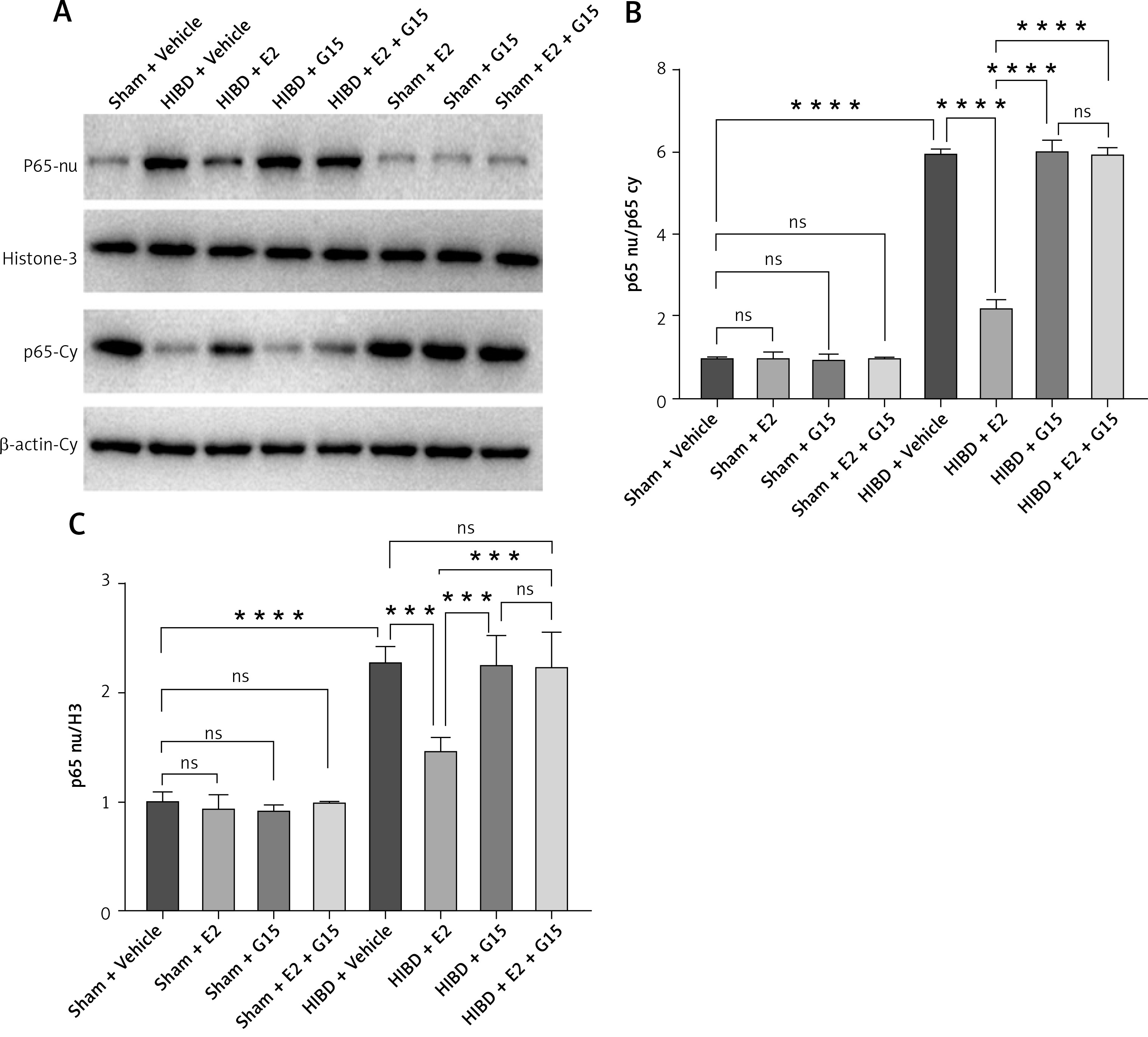
E2 promoted p-AKT expression and reduced NF-κBp65 in ischemic penumbra
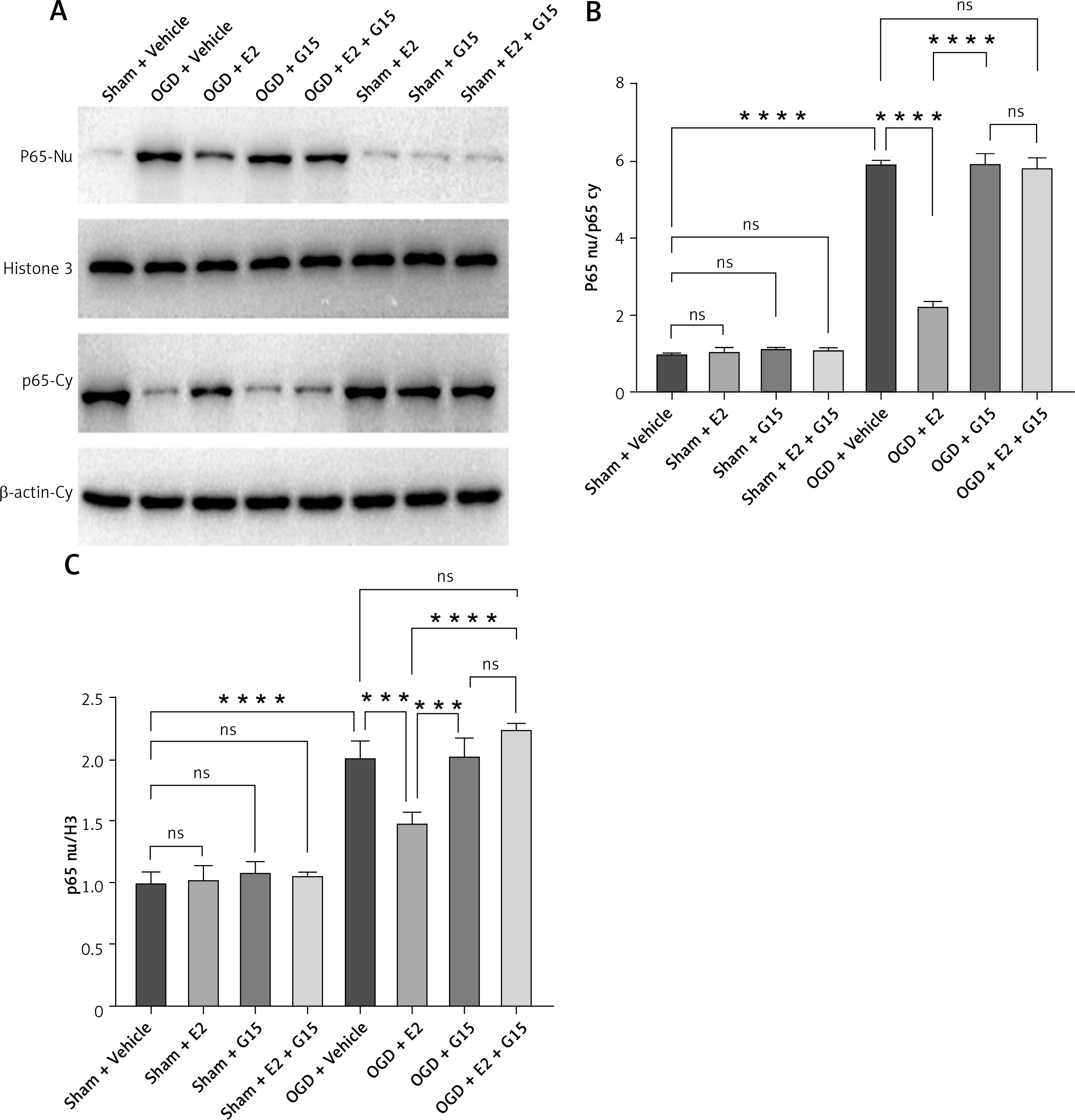
To further investigate whether the GPER1-mediated downstream signaling molecule AKT/NF-κBp65 is involved in E2’s neuroprotective effect on HIBD-affected neonatal rats, we conducted Western blot analysis on ischemic penumbra brain tissue from each group (Figures 4 and 5). The findings revealed that the expression of p-AKT was significantly lower in the HIBD + vehicle group compared to the Sham + vehicle group, mirroring the expression trend of GPER1, while NF-κBp65 expression was significantly higher. Relative to the HIBD + vehicle group, the HIBD + E2 group exhibited notably higher p-AKT expression and markedly lower NF-κBp65 levels. The HIBD + G15 group and the HIBD + E2 + G15 group had significantly lower p-AKT expression and upregulated NF-κBp65 compared to the HIBD + E2 group. No significant differences were observed between the HIBD + vehicle group and the HIBD + E2 + G15 group or between the HIBD + G15 group and the HIBD + E2 + G15 group (Figures 4 and 5).
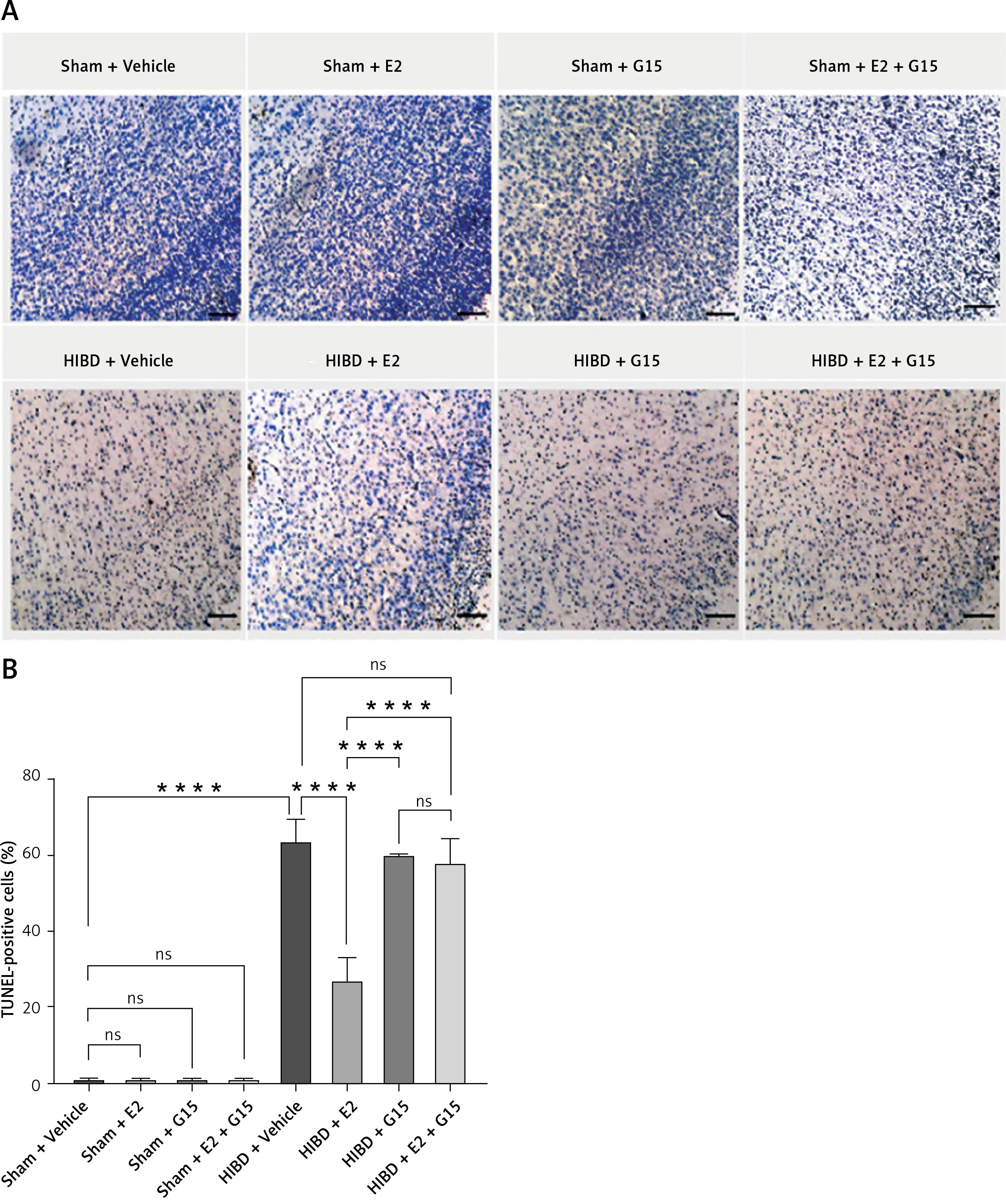
TUNEL staining revealed apoptosis in HIBD rodents
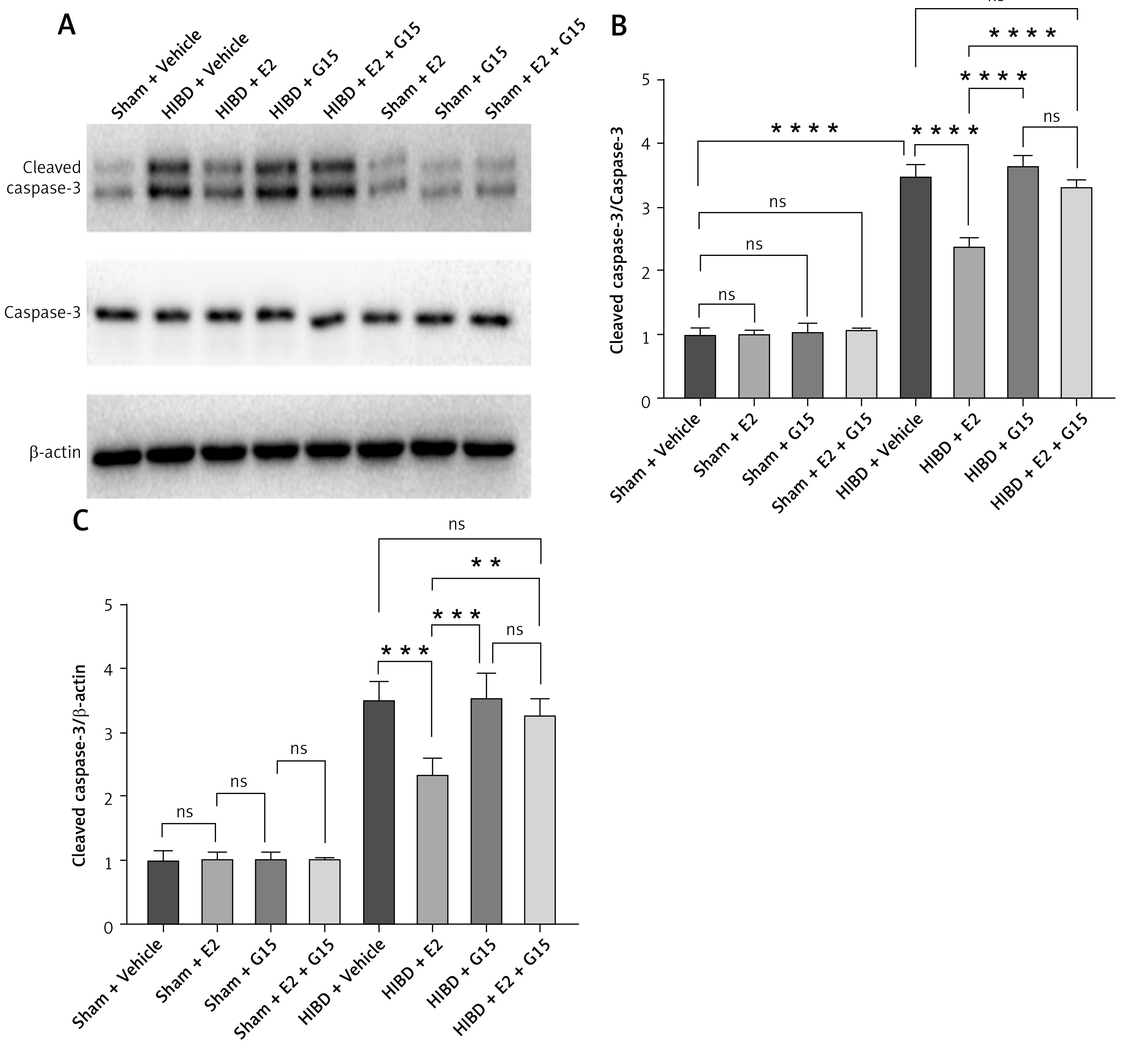
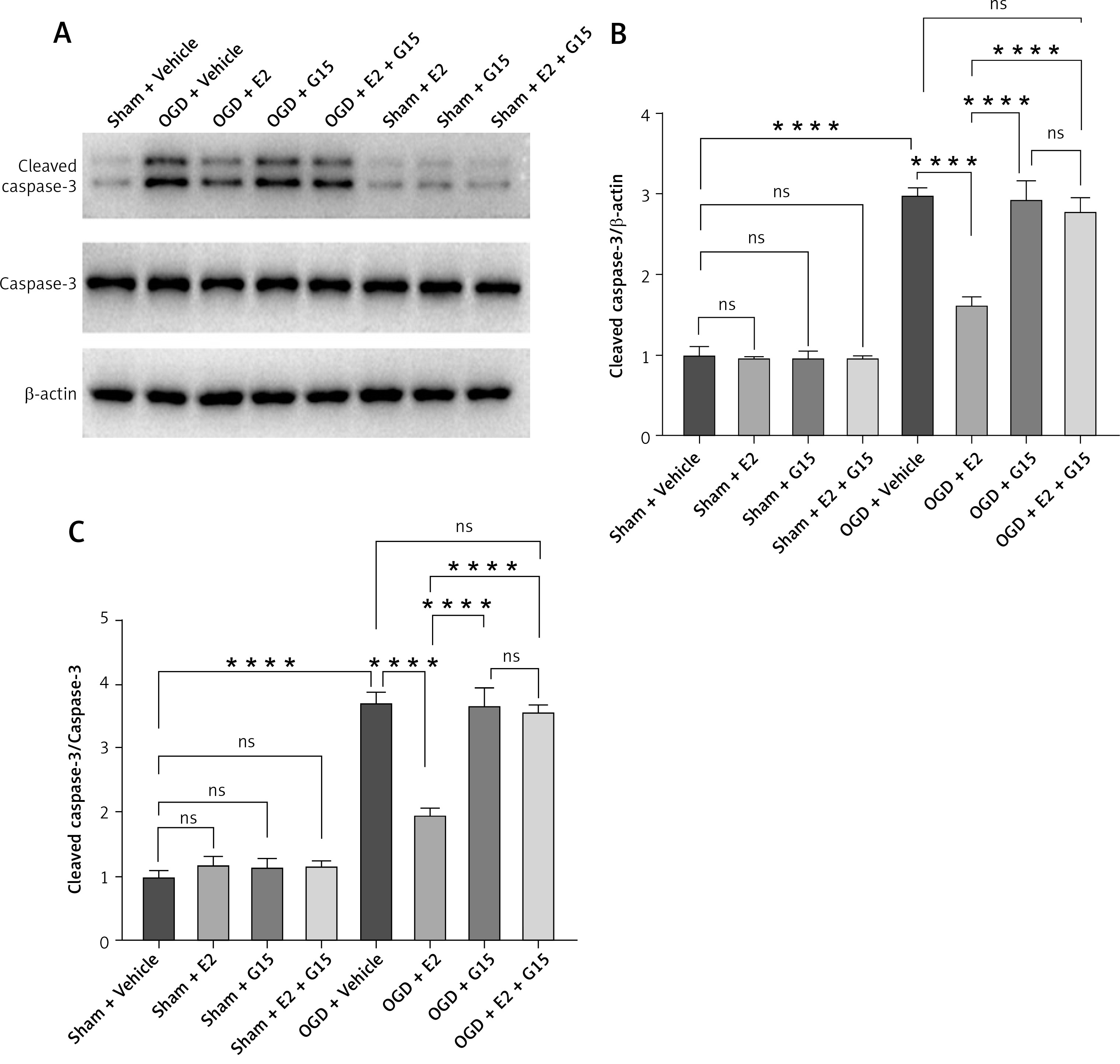
Animal studies utilizing TUNEL staining revealed that the HIBD + E2 group exhibited significantly fewer apoptotic cells within the ischemic penumbra than the HIBD + vehicle group, an effect that was negated following G15 treatment. Apoptotic cell counts in the HIBD + G15 group and the HIBD+E2+G15 group were notably higher than in the HIBD + E2 group (Figure 6). Western blot analysis of cleaved caspase3 paralleled the TUNEL staining results (Figure 7).
Relative cell viability and inflammatory factor expression in OGD astrocytes
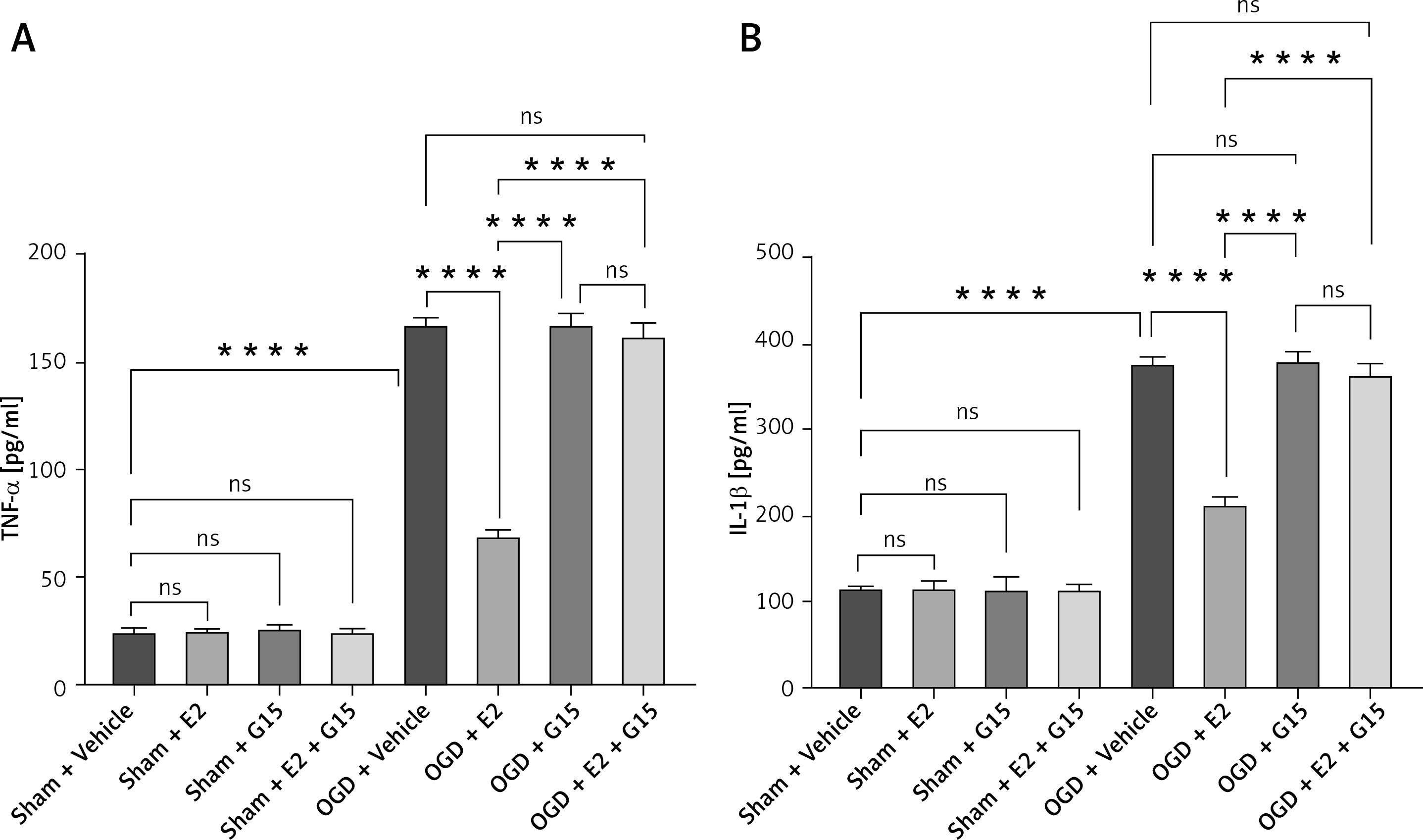
First, we confirmed the expression of GFAP in in-vitro OGD astrocytes (Figure 8). E2 can increase the reduced cell viability in OGD-R astrocytes, while this effect was partly blocked by G15 (Figure 9). E2 could also reduce the increased TNF-α and IL-1β, while G15 could block these inflammatory markers in OGD astrocytes (Figure 10).
Figure 8
GFAP identification of primary cultured astrocytes, n = 5; purity of astrocytes is 97–100%, bar = 100 μm (200×)
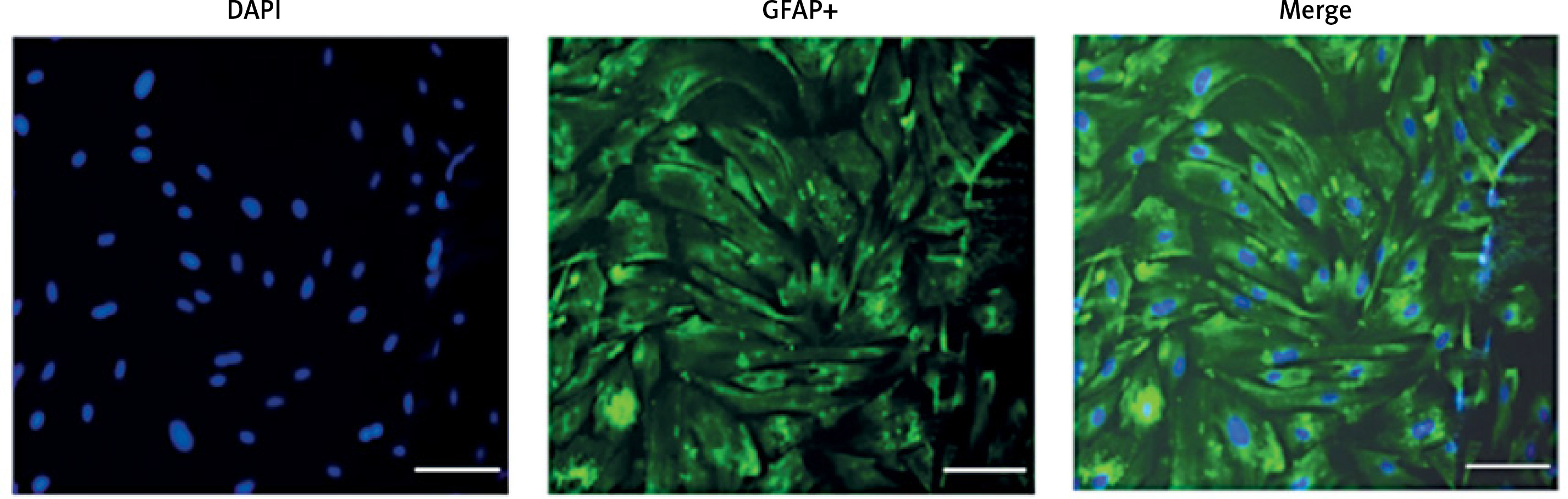
Figure 9
Effects of E2 and G15 intervention on viability of OGD-R astrocytes. N ≥ 5, *p < 0.05, ***p < 0.0005, ****p < 0.0001
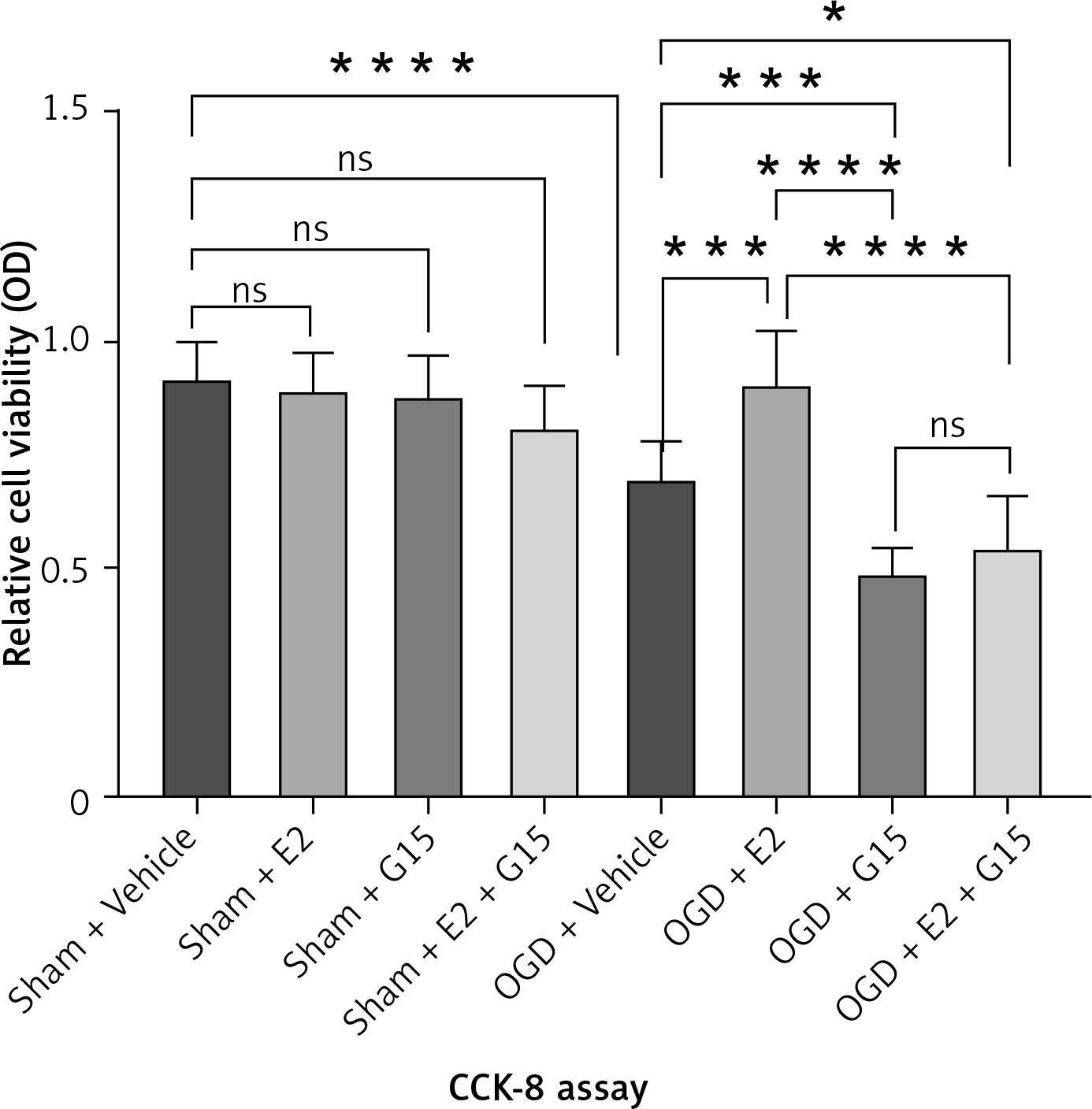
Figure 10
ELISA detected expression of TNF-α and IL-1β in cell supernatant. A – TNF-α; B – IL-1β, n = 5; ****p < 0.0001
Cell experiments mirrored these outcomes, as consistent in the TUNEL staining (Figure 6). E2 could reduce the increased TUNEL-positive cells percentage in OGD astrocyte while G15 could block these effects (Figure 11). Cell experiments mirrored these outcomes, as seen in cleaved-caspase3 expression (Figure 12). E2 could reduce the cleaved-caspase3 level in OGD-R astrocyte while G15 could block these effects.
Figure 11
Effect of E2 and G15 intervention on apoptosis of OGD-R astrocytes. A – TUNEL for evaluating cell apoptosis (200×), bar = 100 μm; B – data analysis; n = 5, ***p < 0.0001
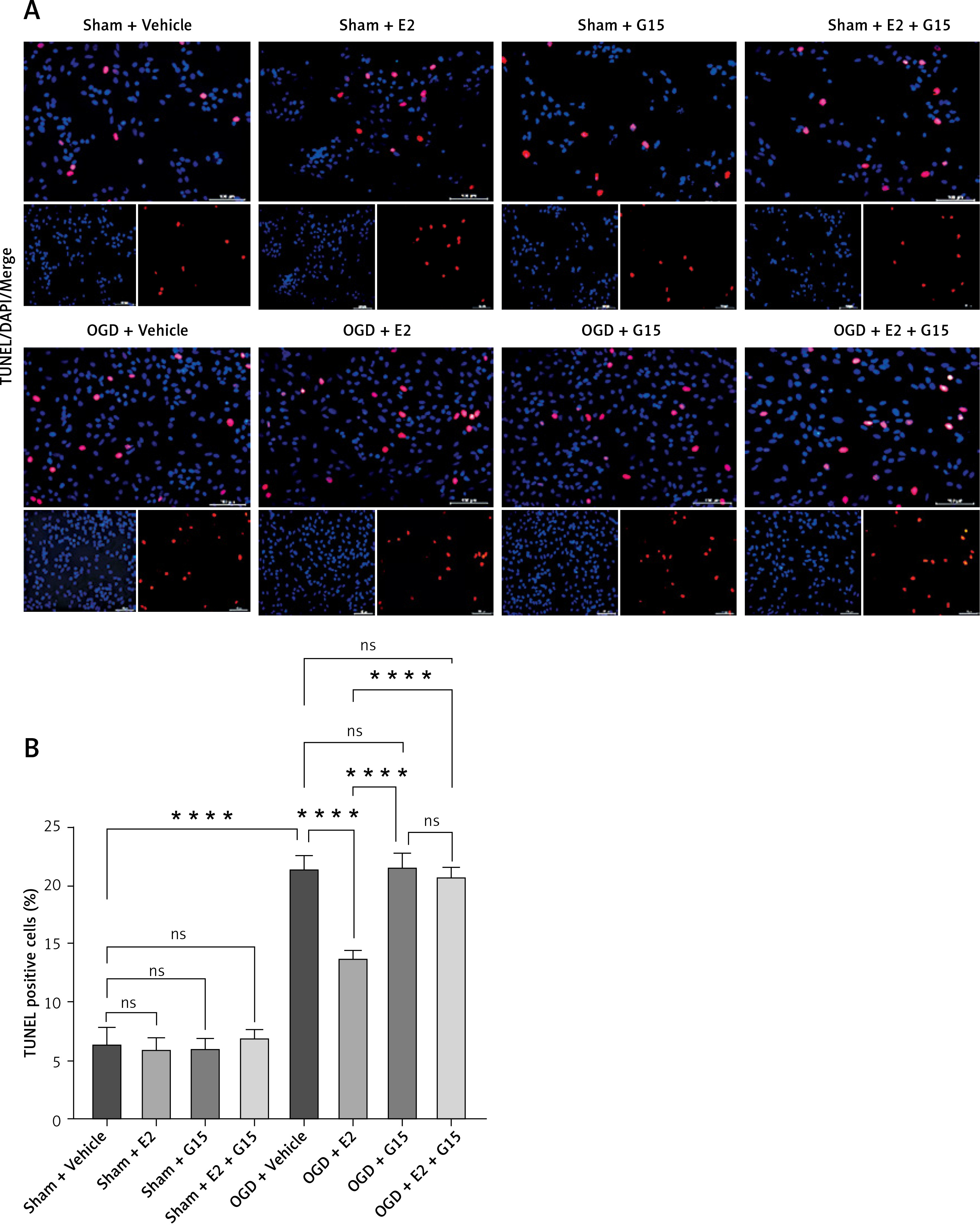
Figure 12
WB detected expression of cleaved caspase-3 in astrocytes after OGD-R. A – representative WB images; B, C – quantification analysis for WB: n = 3, ****p < 0.0001
In vitro experiments showed that the immunofluorescence and WB results for p-AKT and NF-κBp65 were consistent with the in vivo findings (Figures 13–16), suggesting that GPER1 expression decreased on the third post-surgery day in the HIBD group, accompanied by a decrease in p-AKT expression and an increase in NF-κBp65 expression. E2 intervention promoted GPER1 expression (Figure 17) and was accompanied by an increase in p-AKT expression and a decrease in NF-κBp65 expression, which was reversed following G15 treatment.
Figure 13
Effect of E2 and G15 intervention on p-AKT expression in OGD-R astrocytes. A – Immunofluorescence detection of expression of p-AKT (200×), bar = 100 μm; B – data analysis; n = 6, ***p < 0.0001
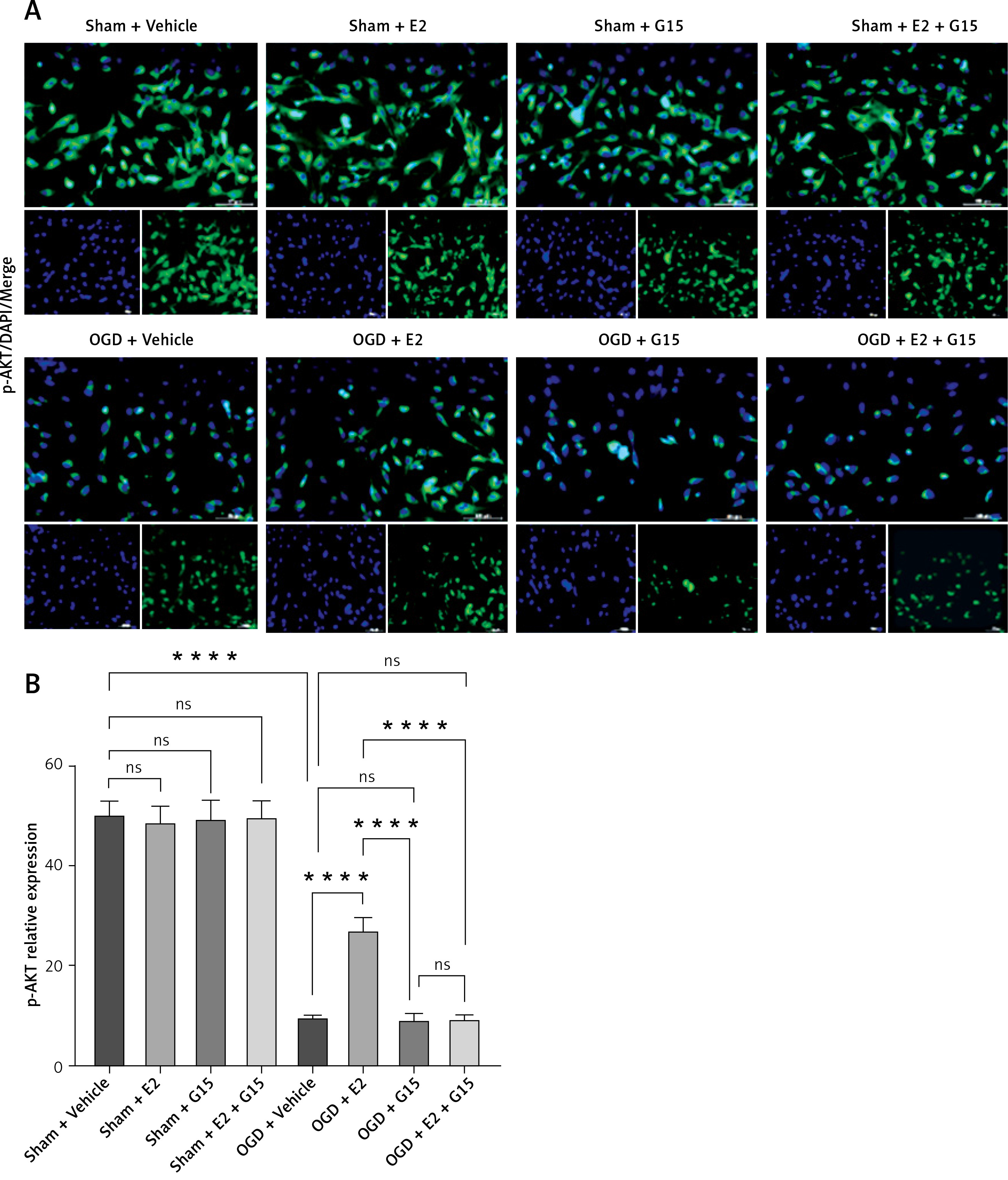
Figure 14
Effect of E2 and G15 intervention on NF-κB p65 expression in OGD-R astrocytes. A – Immunofluorescence detection of expression of NF- κBp65 (200×), bar = 100 μm; B – data analysis, n = 6, ***p < 0.0001
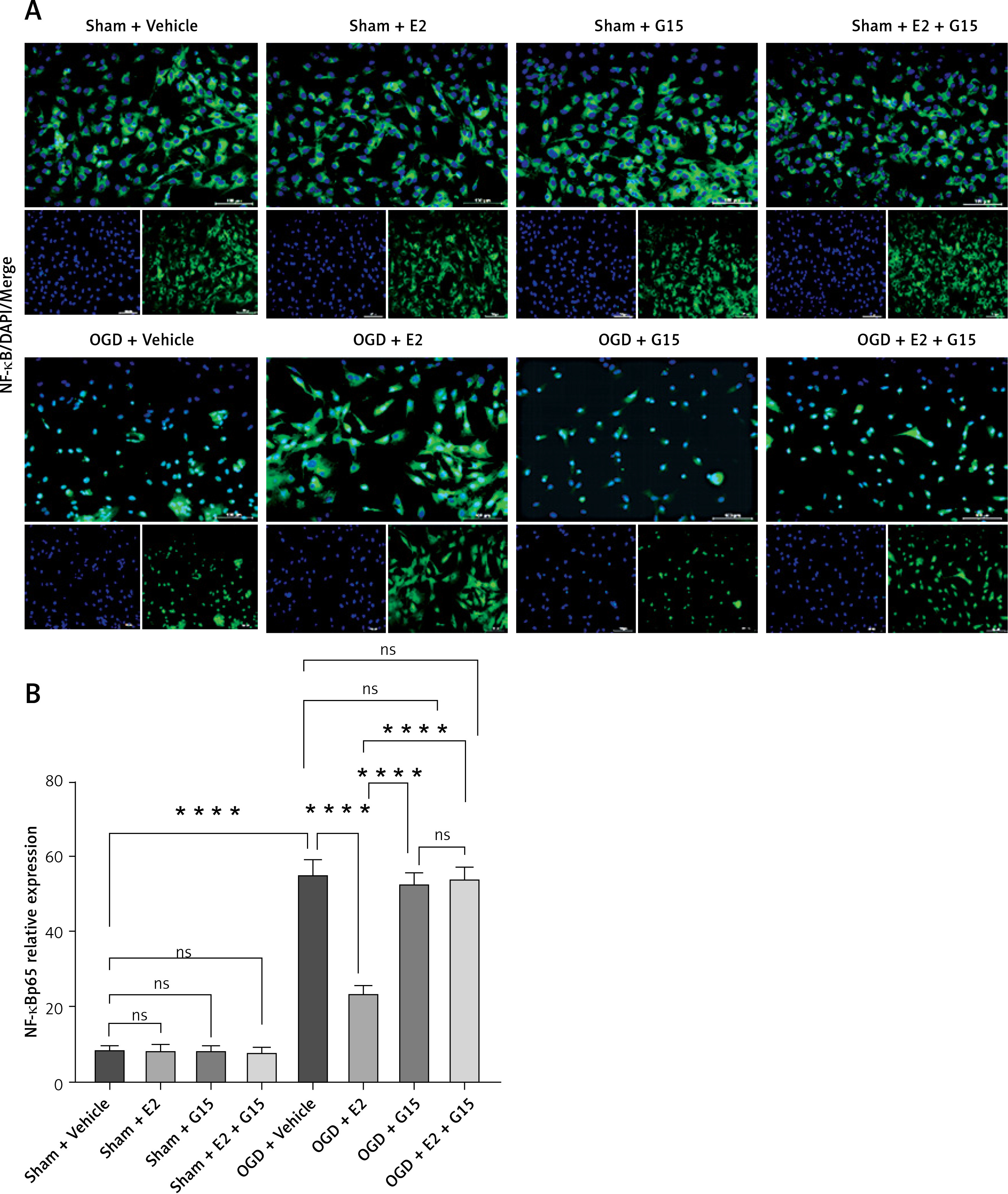
Figure 15
WB detected expression of p-AKT in astrocytes after OGD-R. A – Representative WB images; B, C – quantification analysis for WB, n = 3, ****p < 0.0001
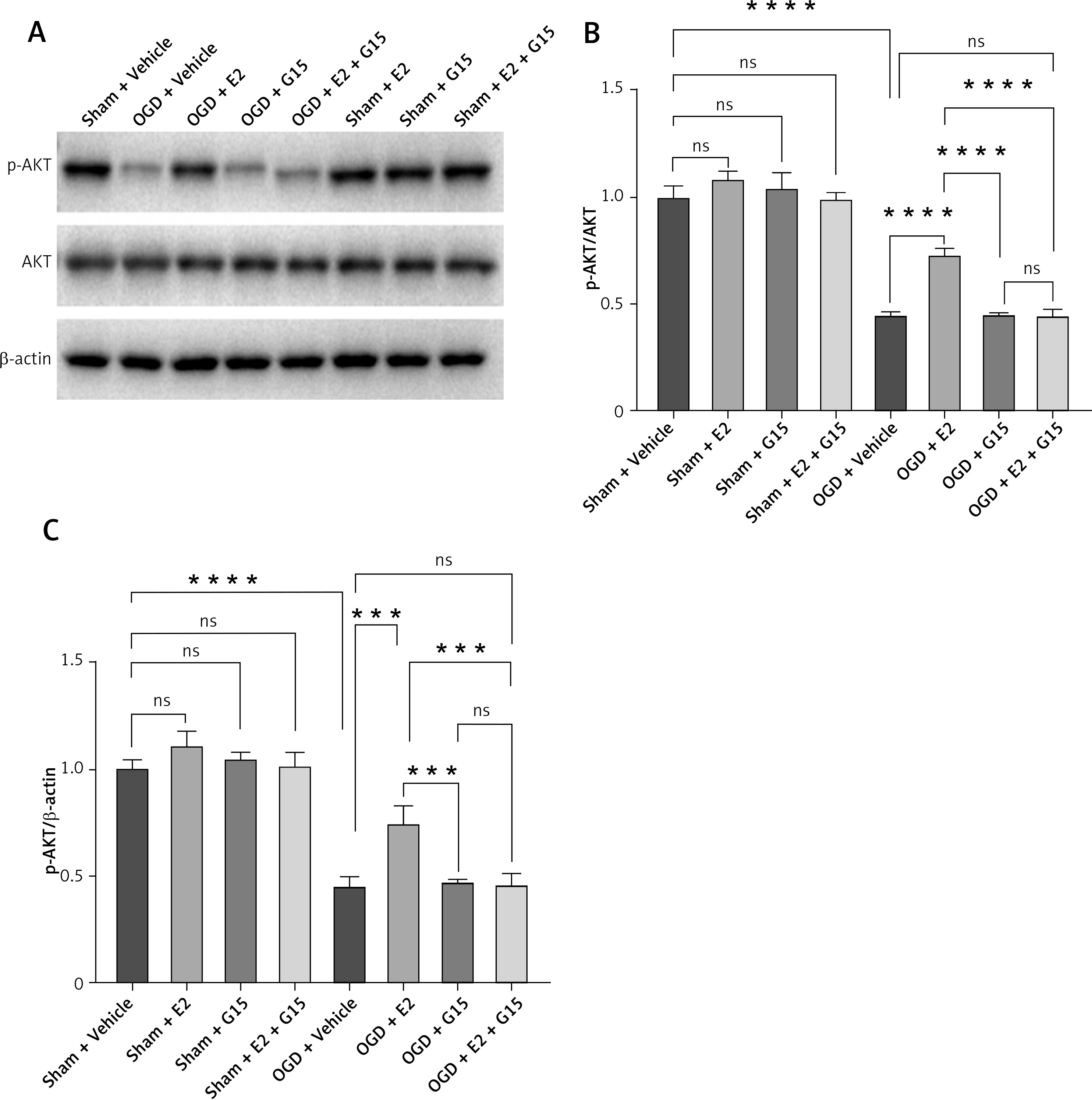
Figure 16
WB detected expression of NF-κBp65 in astrocytes after OGD-R. A – Representative WB images; B, C – quantification analysis for WB, n = 3, ****p < 0.0001
HIBD led to a reduction in GPER1 protein expression in both the ischemic penumbra of neonatal rats and in astrocytes after OGD-R (Figure 17). E2 treatment significantly upregulated GPER1 expression, while G15 effectively inhibited this E2-induced elevation. Astrocyte GPER1 expression after OGD-R reflected these animal experiment results (Figure 17), suggesting that E2 treatment boosts GPER1 levels, which are suppressed by G15. Hence, astrocytes and GPER1 may play a pivotal role in E2’s neuroprotective effect in HIBD-affected neonatal rats.
Discussion
Hypoxic-ischemic encephalopathy (HIE) is a leading cause of neonatal death and long-term neurological disabilities such as movement disorders and cognitive impairments. Current treatments such as mild hypothermia are inadequate, often resulting in permanent neurological damage. There is a pressing need for new therapeutic approaches.
Astrocytes, the most numerous glial cells in the brain, were once thought to exacerbate nervous system diseases when activated [28]. However, recent research highlights their protective roles, such as glutamate clearance, blood-brain barrier repair, and tissue preservation. Enhancing the resilience of glial cells, therefore, represents a promising direction for neuroprotection strategies [29]. After focal ischemia, GFAP knockout mice showed more significant brain damage than their wild-type mice [30]. Treatment with astrocyte conditioned medium after MCAO can reduce the infarct volume and promote the recovery of blood-brain barrier function [31]. Therefore, alleviating the hypoxic-ischemic damage of glial cells may become an important group of neuroprotective strategies.
Previous studies have shown that estradiol has a significant brain protective effect HIBD [32, 33], but its mechanism is still unclear. This study investigated E2’s therapeutic potential in neonatal rats with HIBD and in astrocytes after oxygen-glucose deprivation/reperfusion (OGD-R). In order to explore the possible mechanism of the protective effect of E2 on neonatal rats with HIBD, firstly, we validated the neuroprotective effect of E2 in neonatal rats with HIBD and in astrocytes after OGD-R. So far, four estrogens have been found in the human body: estrone (E1), 17β-estradiol (E2), estriol (E3) and estriol (E4) [34]. “Estrogen” usually refers to E2, because it is widely distributed in many tissues and organ systems and has active physiological functions [35]. Various clinical studies suggest that E2 can prevent or delay the onset of nerve regeneration diseases, such as stroke, seizure, hypoxia and glucose deficiency and oxidative stress, and alleviate damage of the central nervous system [6]. Nuñez et al. found that repeated administration of E2 for three times provided about 70% protection to the hippocampus, basal ganglia and amygdala by constructing the same hypoxic-ischemic model. They believed that E2, as an effective neuroprotective agent, could resist the damage caused by hypoxia-ischemia to the developing brain, and the pretreatment of infants at risk of hypoxic-ischemic damage might have clinical prospects [32]. It has also been reported that E2 can reverse the decrease of neuroprotective astrocyte reactivity in the mouse model of global cerebral ischemia and play a role in brain protection [36]. In vitro studies have also found that E2 replacement therapy can alleviate the cell damage caused by oxygen-glucose deprivation and reperfusion in astrocytes [37]. Previous studies have found that E2 plays a powerful neuroprotective role in the ischemic penumbra [38]. Consistent with the above studies, continuous E2 intervention can significantly reduce the expression level of cleaved caspase-3 and inhibit apoptosis of cells in the ischemic penumbra and astrocytes after OGD-R, thereby reducing the infarct area of brain tissue and improving motor sensory nerve function, achieving neuroprotective effects. However, G15 intervention reversed this protective effect.
We further explored the role of GPER1, an estrogen receptor, in E2’s neuroprotective action. GPER1 is expressed in various brain regions, including the cortex and hippocampus. It mediates estrogen’s protective effects in several contexts, such as reducing infarct volume and inflammatory cytokine release in ischemic models [39]. The neuroprotective effect of E2 was also found in primary hippocampal neurons and rat motor neurons exposed to oxygen glucose deprivation [40]. In addition, GPER mediates glutamate to up-regulate expression of the transporter GLT-1 and the uptake of glutamate by astrocytes, which is involved in reducing excitotoxic nerve damage [17]. In vivo, GPER1 reduced the infarct volume and the release of TNF-α, IL-1β and IL-6 in ischemic penumbra, and participated in the neuroprotective effect of E2 in local and global ischemic models [10]. In this study, we observed that E2 treatment upregulated GPER1 expression, which correlates with reduced infarct size, apoptosis, and improved motor function. Conversely, G15 treatment reduced GPER1 expression and the protective effects of E2, suggesting that GPER1 mediates E2’s neuroprotection in neonatal HIBD.
We also investigated whether GPER1 mediates E2’s neuroprotective effects in astrocytes by primary culture and OGD-R model construction. Research has shown that astrocytes play a crucial role in synaptic transmission and plasticity, and GPER1 activation in astrocytes can mitigate neuronal damage in ischemic conditions. Our results suggest that E2 treatment after OGD-R significantly increases GPER1 expression in astrocytes, reducing apoptosis and inflammatory responses. However, G15 treatment inhibits these protective effects, indicating that GPER1 may mediate E2’s neuroprotection.
We then explored the potential signaling mechanisms behind GPER1’s mediation of E2’s protective effects. GPER1, similar to other G protein-coupled receptors, is responsible for rapid estrogen signal transduction and is independent of the traditional estrogen receptors ERα and ERβ. The PI3K/AKT pathway, a downstream signaling pathway of GPER1, is known to influence cell survival and apoptosis. In ischemic diseases, the activation of GPER1 in astrocytes can restore the basic level of autophagy, reduce the release of inflammatory factors and prevent neuronal damage [41]. In cerebellar development, S-equol in astrocytes may affect neurons and astrocytes through various signal pathways including GPR30. This protective effect can be inhibited by G15 [42].
In the present study, we studied the possible signaling mechanism whereby astrocytes and GPER1 participate in mediating the protective effect of E2 on HIBD in neonatal rats. The structure of GPER1 is similar to other G protein-coupled receptors, and it is mainly composed of α, β and γ subunits, which are responsible for rapid estrogen signal transduction [14]. Although some ERα and ERβ members are also related to plasma membrane and non-genomic signal transduction, the function and expression of GPER1 are independent of these two ERs. GPER1 has high affinity for estrogen and a single binding site [43]. PI3K/AKT is one of the downstream signal pathways of GPER1. Akt participates in cell apoptosis and cell proliferation by influencing the activation state of various downstream effectors. It has been observed in many studies that GPR30 may regulate the PI3K/Akt pathway. For example, it was found that GPR30 activates PI3K/Akt signaling, which is an important step in regulation of neurite formation and protection of cognitive function in the development of hippocampal neurons [44]. It was also found that E2 can activate the PI3K/AKT signaling pathway through GPR30 to play a neuroprotective role in a spinal cord injury model [45]. It has also been reported that the neuroprotective effect of estrogen is related to the rapid activation of Akt and can inhibit cell apoptosis [19].
Finally, we further explored whether NF-κB, a downstream signal molecule of PI3K/AKT, is involved in the protective effect of E2 on the brain of neonatal rats with HIBD, and clarified the relationship between GPER1 and the AKT/NF-κB signaling pathway. NF-κB is a multifunctional transcription factor, which is related to the regulation of many different biological phenomena and disease states, including inflammation, immunity, cell death and the stress response [46]. When cells are exposed to oxidative stress, the PI3K/AKT signaling pathway is activated to regulate the downstream signaling factor NF-κB [20]. Studies show that NF-κB promotes inflammation and apoptosis in cerebral ischemia. So, inhibiting the NF-κB signaling pathway may be a therapeutic strategy for ischemic cerebral infarction [47].
Some studies have suggested that AKT plays a positive role in regulating NF-κB after activation. For example, Song found that Akt plays a neuroprotective role in cerebral ischemia in mice by activating NF-κB [48]. However, some studies have also found that the PI3K-Akt pathway can negatively regulate the expression of NF-κB and inflammatory genes [49]. For example, Li et al. found that G-CSF can reduce neuroinflammation by activating the PI3K/Akt signaling pathway and reducing the expression of inflammatory factors such as NF-κB, TNF-α and IL-1β after hypoxia and ischemia in neonatal rats [50]. Based on the different results of the above studies, we believe that the protective effect of E2 on ischemic brain damage may be affected by many factors. For example, animal species, animal models, degree of brain damage, ways and means of treatment, and even different estrogen levels and sampling times may affect its protective effect and the changes of related receptor expression and signal pathway [51]. Although these studies are contradictory, they show the importance of estrogen receptors in various aspects of ischemic brain damage.
However, several limitations need to be explored in future, such as: whether the agonist G1 of GPER1 has the same neuroprotective effect in neonatal rats with HIBD; what the relationship between GPER1 and classical estrogen receptors is; and whether there is a relationship between the AKT/NF-κB signaling pathway and classical estrogen receptors.
In conclusion, our study suggests that E2 treatment can significantly reduce brain injury and astrocyte apoptosis in neonatal HIBD, with GPER1 playing a central role in mediating these effects through the AKT/NF-κB signaling pathway. Further research is needed to understand the relationship between GPER1 and classical estrogen receptors and the detailed mechanisms of the AKT/NF-κB signaling pathway in mediating estrogen’s neuroprotective effects.