Dysfibrinogenemia is a disease characterized by structural or functional abnormalities of fibrinogen and may be due to genetic or acquired causes [1]. The latter demonstrates higher clinical prevalence and can arise from a multitude of etiological factors, including impaired hepatic synthesis, consumptive coagulopathy in disseminated intravascular coagulation (DIC), autoimmune disorders, neoplastic processes, and infectious pathologies [2].
A 74-year-old woman was admitted to our hematology department due to skin ecchymosis and hypofibrinogenemia (0.8 g/l) persisting for 10 days. Prior to admission, laboratory tests revealed elevated cardiac biomarkers, elevated brain natriuretic peptide (BNP), and renal impairment. Although the coagulopathy, interstitial pneumonia, and secondary infection from the prior admission were treated with antibiotics, plasma, and cryoprecipitate, the patient was discharged with persistent hypofibrinogenemia. Her medical history included sequelae of cerebral infarction with residual hemiparesis and dysphagia leading to recurrent aspiration pneumonia, as well as a history of early-stage lung cancer treated with surgery alone.
Physical examination revealed large ecchymoses in the left lumbar region and bilateral coarse breath sounds. Neutrophilia and elevated inflammatory markers, such as procalcitonin and serum amyloid A (SAA, 207.3 mg/l, reference interval, RI 0–10 mg/l) were observed. Serum perinuclear antineutrophil cytoplasmic antibody was positive (titer: 1 : 100) and anti-cyclic citrullinated peptide antibody (ACPA) > 500 U/ml (RI 0–17 U/ml).
Repeated plasma fibrinogen levels (Sysmex, Kobe, Japan, Clauss fibrinogen assay, RI 2–4 g/l) were critically low (< 1 g/l). The patient received intermittent human fibrinogen concentrate transfusions, yet fibrinogen levels remained between 0.4 and 0.9 g/l. The absence of a relevant family history or personal history of hypofibrinogenemia, combined with the Sanger sequencing results, rule out a congenital form.
Subsequent results ruled out hemophagocytic lymphohistiocytosis, synthetic liver failure, and DIC, with no evidence of progressive thrombocytopenia or prolongation of prothrombin time (PT) or activated partial thromboplastin time (APTT). Substantial elevation of ACPA > 500 U/ml was noted, raising consideration of a diagnosis of rheumatoid arthritis (RA). Given the absence of joint deformities, non-supportive hand radiographs, and a score of 4 on the 2010 ACR/EULAR criteria, the findings do not fulfill the diagnostic requirements for RA. Moreover, positron emission tomography and computed tomography scans showed no tumor recurrence, ruling out paraneoplastic syndrome. Serum protein electrophoresis and immunofixation showed no monoclonal protein, and flow cytometric analysis only found 1.6% reactive plasma cells (CD38++CD138++CD19+CD56-) in bone marrow, excluding multiple myeloma. It is necessary to consider whether anti-fibrinogen antibodies or abnormal fibrinogen may be generated.
The patient’s prolonged thrombin time and low fibrinogen levels were both corrected by the addition of normal plasma, suggesting the absence of anti-fibrinogen antibodies. The most commonly used method for plasma fibrinogen measurement in clinical practice is the Clauss assay, which reflects fibrinogen activity. Fibrinogen antigen level measured by enzyme-linked immunosorbent assay (Shanghai Sun Biotechnology Co., Ltd., Turbidity Immunoassay) was normal (2.8, RI 1.8–3.5 g/l), and the functional-to-antigen ratio was 0.36 (RI > 0.7), indicating dysfibrinogenemia.
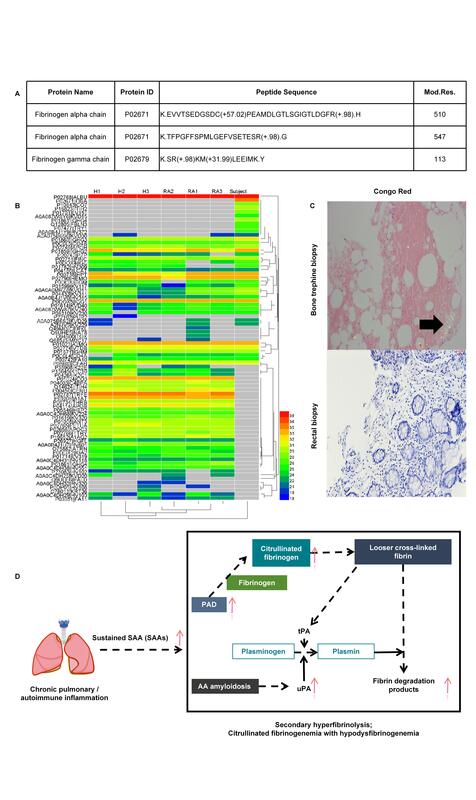
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) revealed widespread protein citrullination, including fibrinogen α and γ chains, confirming the presence of citrullinated-fibrinogen (Figure 1 A). Specifically, two citrullination sites in fibrinogen Aα-chain were detected in our patient (Cit510 and Cit547), and the former site was reported in a previous study [3]. According to the clustering analysis for the area values of citrullinated protein, we found that the type and number of citrullinated proteins in our patient serum were remarkably different from both RA and non-RA controls (Figure 1 B; characteristics are shown in Supplementary Table SI). Moreover, recent studies showed that the composition of fibrin fibers could be altered by fibrinogen citrullination, which leads to a looser fibrin network [4, 5] and increases resistance to lysis in vitro [6]. This suggests that citrullination of the patient’s fibrinogen resulted in a dysfunctional network prone to reduced activity and thereby exacerbated consumption.
Figure 1
Citrullinated proteins and histopathological findings shown in subjects. A – List of fibrinogen-related proteins where citrullination has been annotated directly on the fragment by LC-MS. B – Area values of citrullinated protein measured in serum by LC-MS from rheumatoid arthritis (RA) patients, relatively healthy controls (non-RA, H), and the subject. The Easy-nLC1200-Q-Exactive Plus instrument (Thermo Fisher Scientific) was used, and data were analyzed using the Human_uniprot database (https://www.uniprot.org/, precursor mass tolerance: 10 ppm, fragment ion mass tolerance: 0.02 Da). C – Histopathological results of bone trephine and rectal biopsy stained with Congo red stain; Congo red-positive amyloid-like areas show apple-green birefringence. D – Underlying mechanism and diagnosis
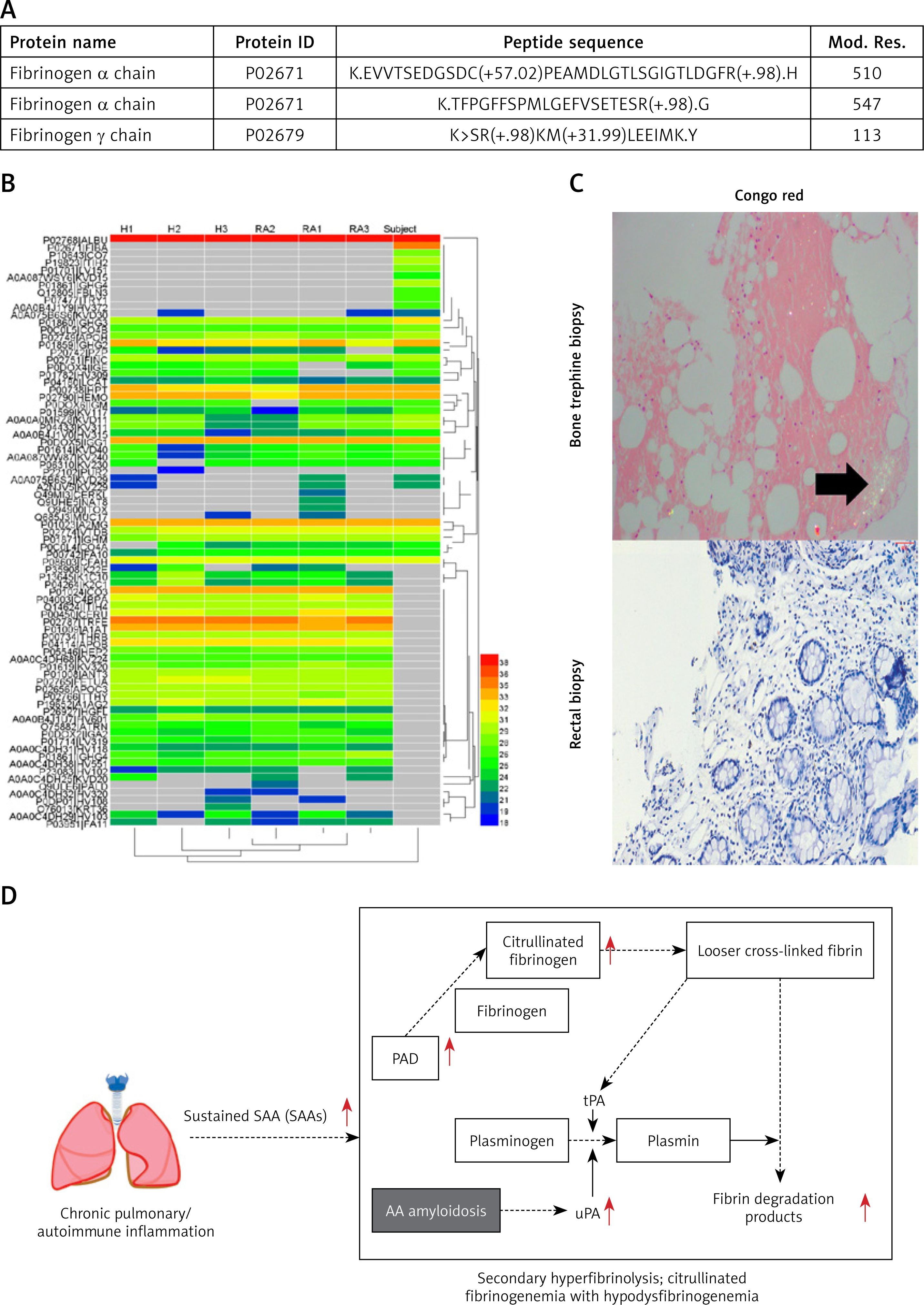
Chronic inflammation and recurrent infections led to sustained SAA elevation, and could develop into AA amyloidosis [7]. SAA was reported to upregulate peptidylarginine deiminase 2/4 (PAD2/4), promoting fibrinogen citrullination [8]. Bone marrow biopsy with positive Congo red staining confirmed AA amyloidosis (Figure 1 C). Additionally, AA amyloidosis may enhance urokinase-type plasminogen activator activity, tilting the balance toward fibrinolysis and further reducing the fibrinogen level [9]. Cardiac and renal dysfunction were also partly attributed to amyloid deposition. The mechanisms described explain the patient’s abnormal fibrinogen and a range of related manifestations (Figure 1 D).
The patient was ultimately diagnosed with interstitial pneumonia with infection, AA amyloidosis, cardiac dysfunction, renal insufficiency, secondary hyperfibrinolysis, and citrullinated-fibrinogenemia with hypodysfibrinogenemia. Targeted antibiotic therapy effectively reduced SAA. Plasma exchange was performed to remove SAA and ACPA, with supplemental fibrinogen infusion. While the therapy did not reverse amyloid deposition, it provided partial improvement. The patient was discharged after infection control and symptomatic improvement.
ACPA is a widely employed diagnostic biomarker for autoimmune disease, especially for RA. In this case, the presence of ACPA served as a critical clue to elucidate the underlying cause of dysfibrinogenemia. The coexistence of ACPA, citrullinated fibrinogen, chronic inflammation, and amyloid deposition suggests a novel pathogenic link. This case suggests that SAA may act as a potential driver of acquired dysfibrinogenemia in the context of chronic inflammation. Furthermore, other molecules such as vascular endothelial growth factor (VEGF) have been shown to play important roles in endothelial dysfunction and accelerated atherosclerosis in chronic inflammatory immune-mediated diseases [10]. Notably, VEGF levels are significantly correlated with fibrinogen levels in RA patients with cardiovascular disease [11, 12]. The patient described herein had a history of cerebral infarction and cardiac dysfunction, but lacked traditional risk factors such as diabetes or hypertension, suggesting that autoimmune dysfunction may lead to endothelial abnormalities, thereby promoting atherosclerosis and vascular disease, ultimately resulting in cerebral infarction and cardiac dysfunction. Given that the concentration levels of VEGF and related molecules were not measured in this study, further validation of the above associations is needed. The case highlighted the clinical spectrum of ACPA and fibrinogen citrullination beyond rheumatoid arthritis and underscores the need for further investigation into its role in systemic inflammatory diseases.