
Introduction
Chronic obstructive pulmonary disease (COPD) is a prevalent irreversible severe respiratory disorder, which is mainly manifested as airflow restriction, resulting in dyspnea, cough, expectoration and other symptoms, seriously affecting the quality of life of patients [1–3]. At present, there are over 300 million COPD patients worldwide, and this number is constantly increasing, imposing a huge burden on individuals and society [4–6]. Chronic obstructive pulmonary disease is a complex disease that is related to various factors such as smoking, environmental pollution, genetics, etc. [7–9]. Research has shown that the onset of COPD involves multiple metabolic and signaling pathways, including inflammatory and oxidative stress responses [10, 11], cell apoptosis, the TNF-α signaling pathway [12], the Wnt/β-catenin signaling pathway [13], etc. In addition, there are some genes related to the genetic mechanism of COPD, including RPTK, interleukin (IL)-6, GSTT1, NRF2, HHIP, and TGF-β. According to some authors [14–16], different variations in these genes may affect the occurrence and development of COPD. Research indicates that chemokines, particularly CXCL8 (IL-8), play a pivotal role in the disease’s pathogenesis. CXCL8 is significantly upregulated in COPD patients, which drives the recruitment and activation of neutrophils to the lung tissues, exacerbating the inflammatory response. This heightened inflammation contributes to the characteristic symptoms and disease progression in COPD.
In addition to chemokines, matrix metalloproteinases (MMPs) are also markedly elevated in COPD. MMPs are enzymes involved in the degradation and remodeling of the extracellular matrix. Their increased activity in COPD patients leads to excessive extracellular matrix remodeling and enhanced cell migration. This results in the breakdown of lung tissue architecture, contributing to the emphysematous changes and loss of lung function observed in COPD patients. These molecular changes highlight potential pathways for therapeutic intervention, aiming to modulate the inflammatory response and prevent tissue destruction in COPD.
The current study leverages advanced transcriptome sequencing and gene expression chip technologies to identify novel molecular markers and therapeutic targets. By comprehensively analyzing gene expression profiles in COPD patients, the study aims to uncover key regulatory molecules involved in disease progression, offering new insights for targeted therapy development [17].
Transcriptome sequencing (RNA-Seq) is a high-throughput sequencing technology used to analyze the expression and structural characteristics of all RNA molecules in samples, which can help researchers understand important biological processes such as gene transcriptional regulation and signaling pathways. RNA-Seq technology has been widely used to study the molecular mechanism of COPD and to identify potential biomarkers and therapeutic targets. Research has shown that compared with healthy control groups, COPD patients exhibit different gene expression patterns and signaling pathway abnormalities, which are closely related to typical characteristics of COPD such as inflammatory response, cell apoptosis, and oxidative stress.
This study combines transcriptome sequencing and microarray expression data to further explore the molecular mechanism of COPD. Through differential analysis, we screened and processed some genes closely related to the pathogenesis of COPD, and combined them with known functional genes and protein interaction networks in the literature to explore potential regulatory genes. Ultimately, we discovered regulatory components that are crucial to the etiology and control of COPD, providing new potential molecular mechanisms for the diagnosis and emergence of therapeutic drugs for COPD.
Material and methods
Data acquisition
GSE57148 used RNA-Seq technology to analyze the gene expression profiling information of lung tissue samples. The data set measured the lung tissue RNA transcription data of 98 COPD subjects and 91 healthy control samples using HiSeq200 platform [18]. GSE38974 measured gene expression data of miRNAs and mRNA in lung tissues of COPD subjects and control samples based on the GPL4133 and GPL7723 platforms, and explored potential biological regulatory pathways in the pathogenesis of COPD by constructing an mRNA-miRNA interaction network [19]. GSE42057 conducted genome-wide gene expression analysis on 42 healthy control samples and 94 exceptional peripheral blood samples with varying degrees of COPD using the GPL570 platform [20]. Protein interaction information was obtained from the STING (Version 11.5) database, which includes experimentally confirmed protein interaction pairs and method predicted interaction pairs [21].
RNA-Seq data analysis
Using the FastQC (v0.11.9) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) tool we performed quality control of sequencing data, using cutadapt (v4.0) to remove connector sequences and filter low-quality bases. The filtered data were compared to the human reference genome of the hg19 version using TopHat (v2.1.2), and then the transcript abundance information was calculated using Cufflinks (v2.2.1), which were transformed into FPKM (fragments per kilobase of script per million reads mapped) statistics [22]. After standardization of data, limma (v2.10.7) [23] was used for screening differential genes. Considering the size of the sample and referring to other literature indicating a larger number of differential genes, the statistical significance was set as |logFC| > 0.55, p < 0.05. These thresholds were chosen to balance the identification of a meaningful number of differentially expressed genes (DEGs) with statistical rigor. For RNA-Seq data, a logFC threshold of 0.55 was selected to capture genes with moderate expression changes, which are often biologically relevant in COPD pathogenesis.
Chip data analysis
The dataset was downloaded from GEO using GEOquery (https://www.ncbi.nlm.nih.gov/geo/), while integrating platform annotation information. This was followed by merging expression data using Gene Symbol from annotated information and replacing duplicate data with mean, and finally using limma [23] for differential gene mining and screening. The statistical significance setting was |logFC| > 0.89, p < 0.05. We used ggplot2 [24] to visualize gene expression heatmaps, volcanic maps, etc. The chosen thresholds were determined to ensure the detection of genes with substantial expression changes, balancing sensitivity and specificity. This choice reflects the typically higher baseline variability in microarray data compared to RNA-Seq data.
Functional enrichment analysis
To screen differentially expressed genes, clusterProfiler (v4.12.0) [25] was used for Gene Ontology (GO) and KEGG functional enrichment analysis and pathway annotation analysis, and the annotation results were visualized and analyzed.
RT-PCR and expression analysis
For RT-PCR, the total RNA extracted using triReagent was quality checked using spectrophotometry and gel electrophoresis. Following this, the precalculated amount of sample was added with either beta-actin, cytochrome P450 family 1 subfamily B member 1 (CYP1B1) or pentraxin 3 (PTX3) TaqMan probes in an RT-PCR master mix. A one step-reverse transcription PCR was followed. According to the cDNA synthesis program, the first incubation was carried out at 42°C for 15 min. Then a subsequent incubation at 95°C for 5 min followed by continued RT-PCR was carried out using TaqMan probes and primers as specified in supplementary material, using the Bio-Rad CFX96 platform to ascertain the transcription of various genes. The thermal protocol was used, which included a 30-second incubation at 98°C for enzyme activation and 40 cycles of 15 s at 95°C and 1 min at 60°C. The technique incorporated a temperature ramp from 65°C to 95°C with 0.5°C per second increments after the final reaction cycle to omit non-specific products with melting curve data. Numerous housekeeping genes (actin) were used to standardize the data before being compared using ΔΔCt (difference between ΔCt values, which results from the difference between the cycle threshold value Ct of target and housekeeping genes). The results were presented as the fold change in the transcript normalized to housekeeping genes.
Immunoblotting
Anti-CYP1B1, anti-PTX3 and anti-tubulin primary antibodies were used for immunoblotting. Nitrocellulose membrane (cat# Z613657, Sigma, USA) was then treated with blocking solution overnight at 4°C after proteins were separated using SDS-PAGE and transferred to the nitrocellulose membrane utilizing semidry western transfer apparatus (GE Healthcare, USA). On the following day, blots were incubated with the primary antibody for 2 h at a dilution of 1 : 2000, then with the secondary antibody for 1 h at a dilution 1 : 10 000. Each step was preceded by washing three times with phosphate-buffered saline with Tween (PBST) for 5 min. Finally, the blots were developed using chemiluminescent peroxidase substrate and signals were obtained on photographic film (Kodak, XBT film, India). The Gel Documentation System was utilized to capture the images. Utilizing the image analysis program Image J (NIH), the relative densities of the protein bands were calculated and reported as the number of pixels per group.
Results
Results of transcriptome sequencing analysis of COPD
According to the evaluation results of FastQC, the samples were filtered for quality control. TopHat was used to map the RNA-Seq sequence data of qualified samples to the human genome (hg19) and transcript reference, and Cufflinks software was used to calculate the number of gene transcripts and convert them into FPKM to measure the relative abundance of transcripts. We excluded genes with an FPKM value of 0 from all samples. After standardization of the filtered data, we used the limma tool for differential gene identification and screening. The screening conditions are set to |logFC| > 0.55, with a p value < 0.05.
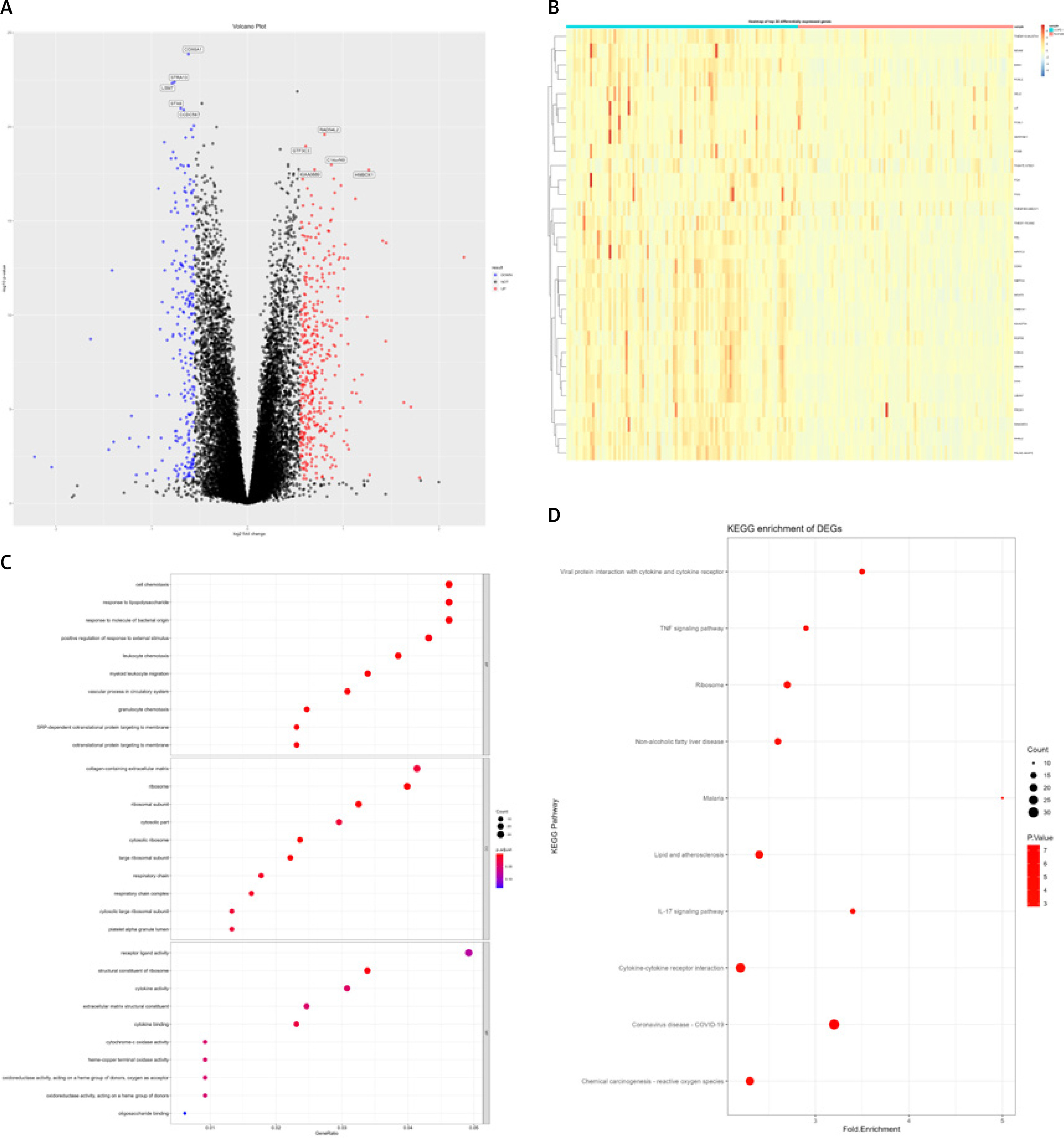
After analysis, a total of 850 differentially expressed genes were selected, including 712 differentially expressed protein coding genes and 138 non-coding genes (Figures 1 A, B). Gene Ontology function enrichment analysis of 712 differential protein coding genes showed that these differential genes were involved in the regulation of cell migration and chemotaxis, including leukocytes, granulocytes and other important cells. At the same time, they also participate in some important biochemical processes related to proteins, lipids, and polysaccharides, and are related to important regulatory processes such as positive regulation of DNA binding transcription factor activity and positive regulation of the response to external stimuli (Figure 1 C). Functional analysis of the KEGG pathway showed that these differential genes are mainly closely related to important signaling pathways such as the IL-17 signaling pathway, PI3K/Akt signaling pathway, TNF signaling pathway, chemokine signaling pathway, the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), VEGF signaling pathway, NOD like receptor signaling pathway, and Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway, and also participate in cytokine-receptor interaction, oxidative phosphorylation, focal induction, cell induction and other important biological processes (Figure 1 D).
Figure 1
Chronic obstructive pulmonary disease (COPD) RNA-Seq data (GSE57148) differentially expressed genes (DEGs) and functional annotation. A – Volcano plot, where blue dots represent low-expressed genes and red dots represent high-expressed genes. B – Heat map of the top 30 differentially expressed genes. C, D – DEGs’ Gene Ontology (GO) functional enrichment analysis (C) and KEGG pathway annotation (D); the size of dots represents the number of enriched genes, and the color of dots from blue to red represents the statistical significance
COPD lung tissue microarray analysis
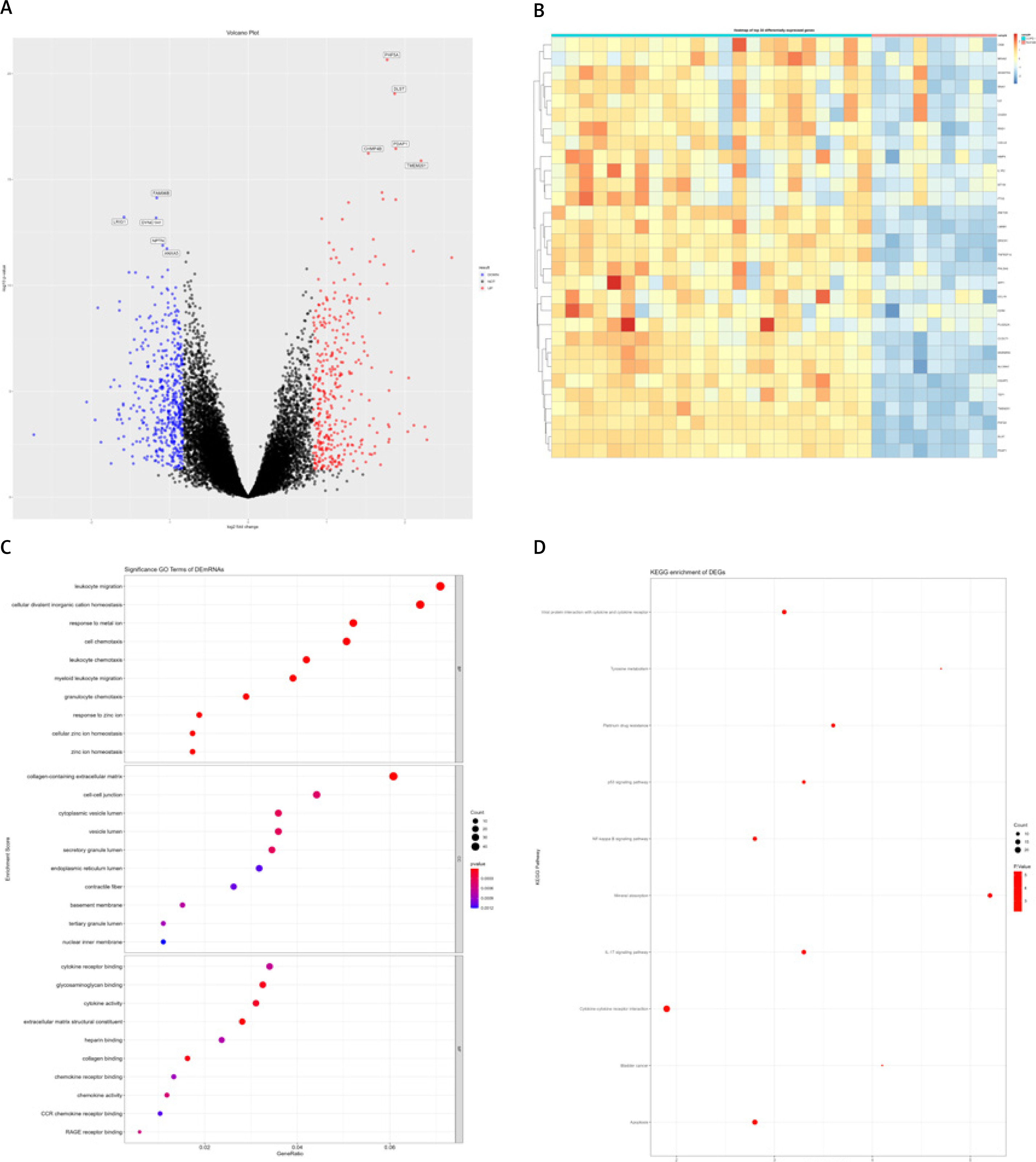
mRNA expression information of lung cancer tissues from COPD and healthy control groups was extracted from the GSE38974 dataset. This analysis included 32 samples, including 23 COPD subjects and 9 healthy control samples. After differential analysis, a total of 828 differentially expressed genes were identified (Figures 2 A, B). Gene Ontology function enrichment analysis of the obtained differential genes found that these differential genes were involved in important cell regulation processes such as cell chemotaxis, cell migration and cell response to the outside world, including leucocyte migration, granulocyte cell migration, and neutrophil cell migration, regulation of leukocyte chemotaxis migration, monocyte migration, cellular response to cadmium ion, and cellular response to zinc ion. In the process of COPD, abnormal expression of multiple molecules will cause inflammatory reaction in the lung, attracting immune cells, including neutrophils, monocytes and lymphocytes to gather in the lung tissue. In these complex regulatory processes, oxidative stress or cytokine stimulation can promote the migration of epithelial and stromal cells. In addition, GO functional enrichment analysis also showed that these differentially expressed genes are involved in cytokine signaling regulation closely related to cellular biological processes, such as negative regulation of growth, the intrinsic apoptotic signaling pathway, cytokine secretion, and positive regulation of cell adhesion. The chemokine-mediated signaling pathway and other multiple factor signaling regulatory processes are mediated by chemokines (Figure 2 C). Functional annotation of differentially expressed genes in the KEGG pathway revealed that these differentially expressed genes are involved in the IL-17 signaling pathway, apoptosis, the p53 signaling pathway, the NF-κB signaling pathway, and TNF (Figure 2 D).
Figure 2
GSE38974 differentially expressed genes (DEGs) and functional annotation. A – Volcano plot, where blue dots represent down-regulated genes and red dots represent up-regulated genes. B – Heat map of the top 30 DEGs. C, D – DEGs’ Gene Ontology (GO) functional enrichment (C) and KEGG pathway annotation (D); the size of dots represents the number of enriched genes, and the color of dots from blue to red represents the statistical significance
Analysis of PBMC chip for COPD
The GSE42057 dataset contains genome-wide expression data from 136 COPD subjects and healthy controls, as well as peripheral blood samples. Differential analysis was performed on these data, and a total of 351 differentially expressed genes were selected, including 305 protein coding genes and 56 non-coding genes. Gene Ontology functional enrichment analysis of these differentially encoded genes revealed that they are involved in the regulation of immune cell differentiation and related signaling pathways, including T cell differentiation, lymphocyte differentiation, T cell activation, and regulation of cell adhesion. The regulation of leukocyte differentiation, the involvement of T cell differentiation in the immune response, and the regulation of IL-12 production are important cellular biological processes related to immune regulation, as well as signal regulation in these processes. It includes the immune response activating the cell surface receptor signaling pathway, antigen receptor mediated signaling pathway and other signaling pathways. The KEGG pathway analysis of differential genes found that these genes were involved in important signal regulation pathways such as the NF-κB signaling pathway, JAK-STAT signaling pathway, T cell receptor signaling pathway, and NOD like receptor signaling pathway. They also participate in Th17 cell differentiation, Th1 and Th2 cell differentiation, cytokine receptor interaction and other important cell processes.
Integrated analysis of genes related to COPD
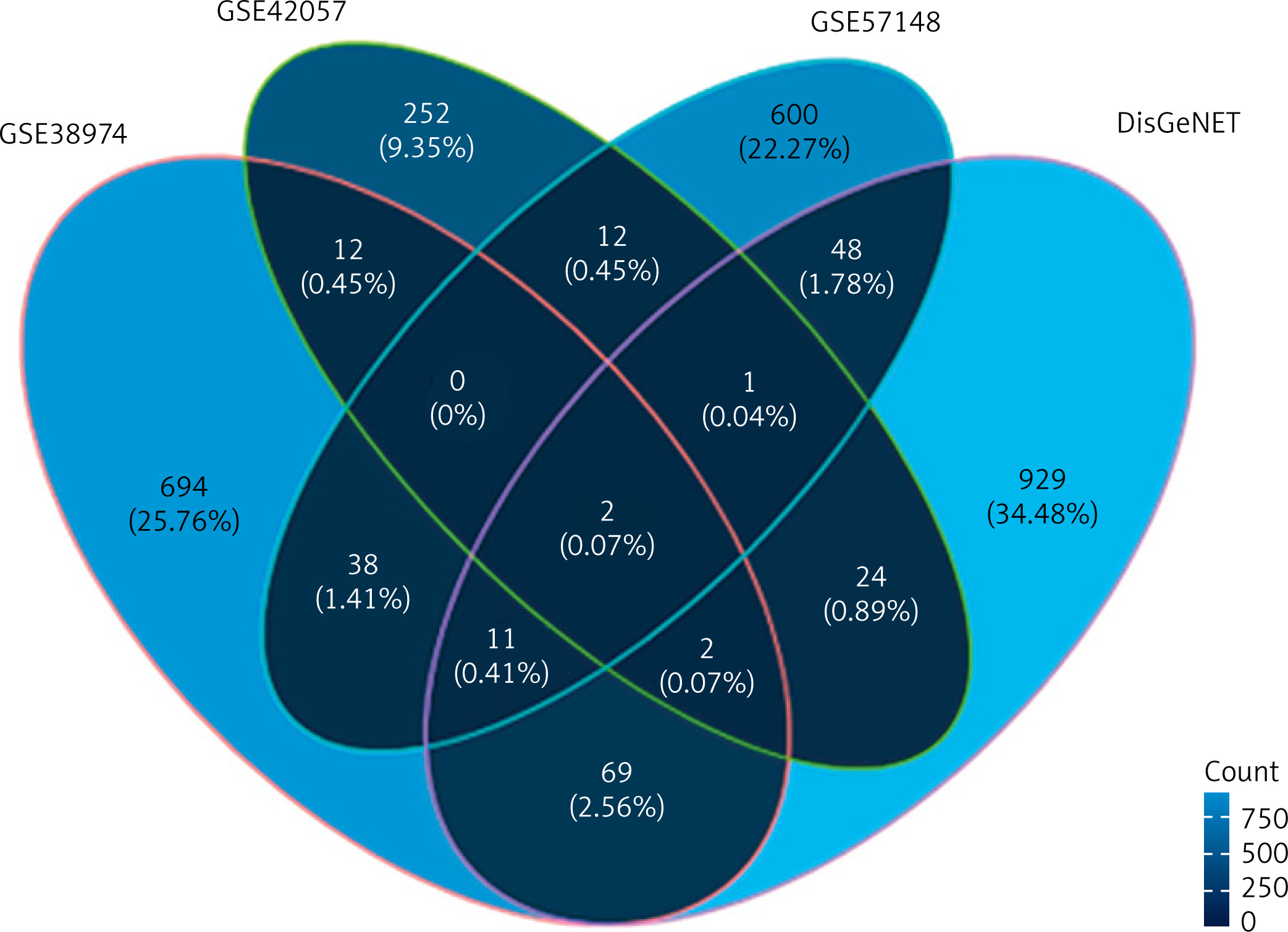
In order to screen for gene markers closely related to COPD function, we investigated this marker’s expected differential expression in COPD lung tissue and peripheral blood. Firstly, we used the RNA-Seq analysis results and chip data analysis results of lung cancer tissue to screen differentially expressed genes related to COPD in lung cancer tissue. This time, a total of 51 differentially expressed gene intersections were screened. Then, the expression of these differential genes was detected in the peripheral blood data of COPD, in order to screen for marker genes related to COPD function using DisGeNET [26] (https://disgenet.org/). The gene expression data related to COPD were retrieved. A total of 1192 genes related to COPD were integrated in this study, including 1086 protein coding genes and 106 non-coding regulatory genes. The intersection of differentially expressed genes and known genes related to COPD was initially analyzed and presented as a Venn diagram, showing only 2 genes common across all the datasets; 694 unique genes in the dataset GSE38974, 252 unique genes in the dataset GSE42057, 600 unique genes in the dataset GSE57148 and 929 unique genes in the DisGeNET dataset were observed (Figure 3).
Protein interaction network analysis
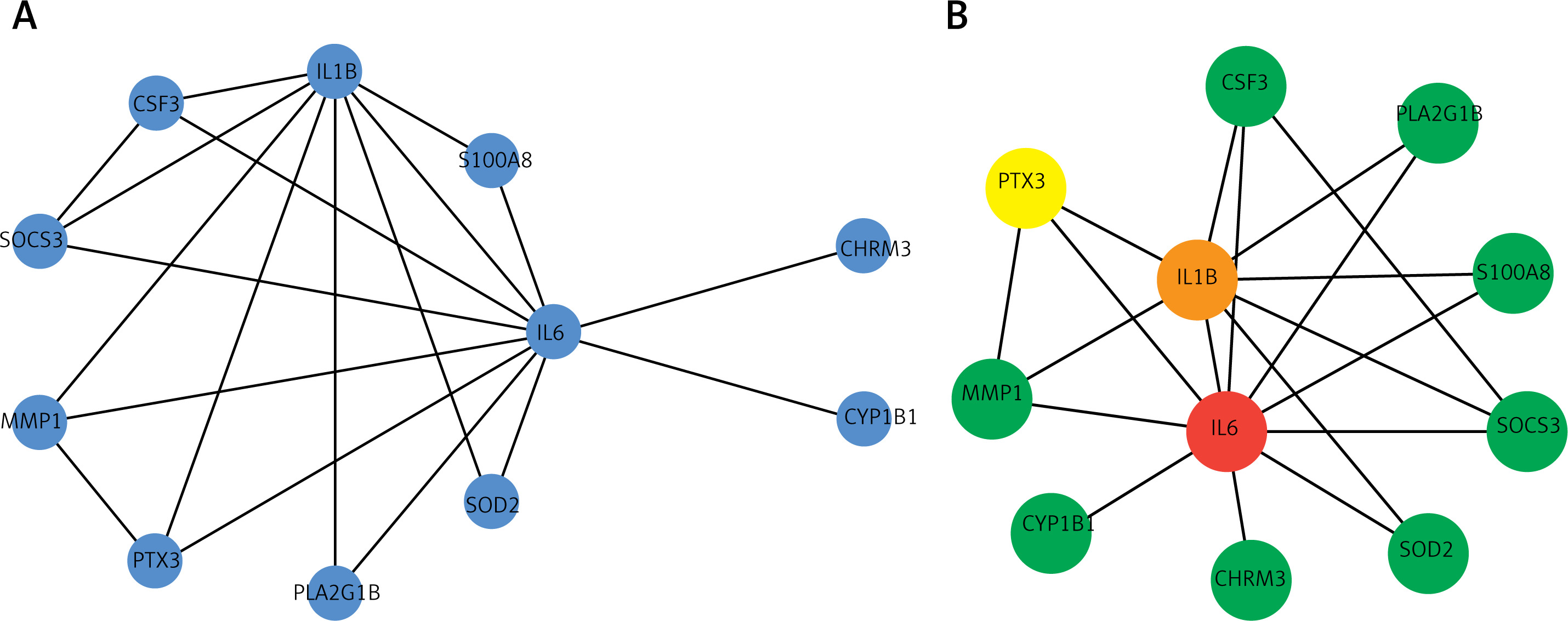
Visualization of the interaction network was performed using Cytoscape [27], in order to evaluate the protein network of the differentially expressed genes in COPD. We further used CytoHubba [28] for module mining. Through network analysis, it was found that IL6, IL1B, and PTX3 rank the highest in the network, indicating that these genes play an important regulatory role in the pathogenesis and development of COPD (Figure 4). Functional enrichment analysis of intersection genes revealed that these genes are involved in cell taxis and metastasis, as well as related signaling and metabolic regulatory pathways. KEGG pathway analysis also found that these important genes are closely related to cell differentiation and signaling and receptor pathways. The IL-17 signaling pathway, TNF signaling pathway, and JAK-STAT signaling pathway have been confirmed in previous studies [10, 13, 16].
Figure 4
Protein interaction network and significant modules of intersection of differentially expressed genes (DEGs) with chronic obstructive pulmonary disease (COPD)-related genes. A – Protein interaction network of intersection of DEGs with COPD-related genes. B – Highly connected module of network A; color from blue to red represents the importance of the node in the network
Gene expression validation
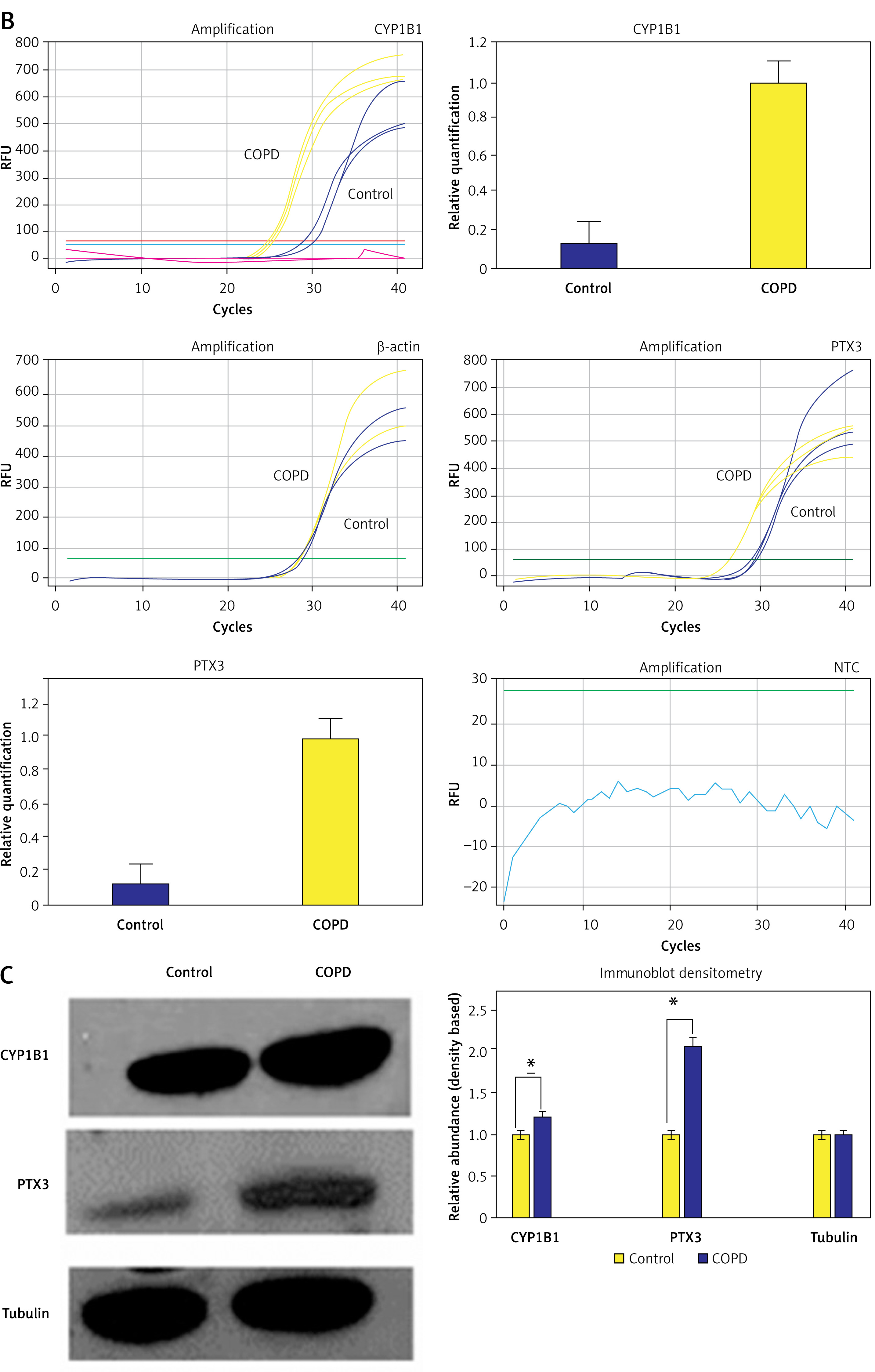
The analysis suggested that the genes CYP1B1 (cytochrome P450 family 1 subfamily a polypeptide 1) and PTX3 (pentaxin related protein PTX3) may play important regulatory roles in the pathogenesis of COPD. We used the data set GSE42057 to verify the detection and expression of these two genes, and found that CYP1B1 and PTX3 were significantly differentially expressed in COPD and control groups (Figure 5 A). Furthermore, to confirm the transcript and translation level changes in COPD with respect to healthy controls, we analyzed the extracted RNA and protein fraction from healthy subjects and COPD plasma. We estimated the transcript using RT-qPCR and further confirmed the protein levels of CYP1B1 and PTX3 using immunoblotting. Using the DDCt method, we observed that the CYP1B1 transcript was 25.3-fold higher in the COPD sample compared to the healthy control and PTX3 was upregulated 8.2-fold in COD samples in contrast to the healthy control (Figure 5 B). Furthermore, immunoblotting and densitometric analysis using ImageJ revealed that the CYP1B1 protein level was 1.22-fold higher in the COPD sample compared to the healthy control. Similarly, PTX3 expression was 2.05-fold upregulated in COPD samples compared to the healthy control (Figure 5 C). The subtle differences in the expression profile observed via qRT-PCR and immunoblotting suggest a strong possibility of post-transcriptional regulation and perhaps regulation via small RNA, which is subject to further exploration. The above experimental findings were concurrent with the large RNA-Seq dataset and corroborate our network analysis confirming the potential of CYP1B1 and PTX3 as strong diagnostic as well as putative therapeutic targets, which need further exploration.
Figure 5
Validation of expression of CYP1B1 and PTX3. A – Expression status of CYP1B1 and PTX3 in dataset GSE42057; CYP1B1 is significant in GSE42057 with different groups, PTX3 is significant in GSE42057 with different Groups. B – qRT-PCR based expression analysis. C – Immunoblotting based protein level analysis
Discussion
Chronic obstructive pulmonary disease is a severe progressive lung disorder characterized by airway inflammation and remodeling, leading to significant morbidity and mortality. This study aimed to identify potential molecular markers and therapeutic targets for COPD by integrating transcriptome-sequencing (RNA-Seq) and gene-expression chip data. Our findings revealed that the NF-κB and JAK-STAT signaling pathways are persistently activated in COPD, driving chronic inflammation and tissue remodeling. Through differential gene analysis, we identified 850 differentially expressed genes, including key regulators such as IL6, IL1B, and PTX3. Notably, PTX3 and CYP1B1 emerged as potential biomarkers and therapeutic targets due to their roles in inflammation, oxidative stress, and tissue remodeling. These discoveries provide new insights into COPD pathogenesis and open avenues for personalized management and novel therapeutic strategies.
The pathogenesis of COPD is subtle and complicated, and its pathogenesis has not been fully elucidated yet. Currently, research suggests that COPD is closely associated with smoking, environmental pollution, and genetic factors, among which smoking is the most important inducing factor [29–32]. The molecular mechanism of COPD involves multiple complex biological processes and regulatory pathways, including the inflammatory response, oxidative stress response, cell apoptosis, cell proliferation, cell chemotaxis, lung tissue remodeling, and fibrosis [33]. These biological processes are related to specific signaling pathways, such as the TNF signaling pathway, redox signaling pathway, IL-17 signaling pathway, cell cycle regulation, and lung tissue remodeling related pathways [34–36].
The NF-κB signaling pathway is a pivotal regulator of immune and inflammatory responses. In COPD, NF-κB is persistently activated, leading to the chronic recruitment and activation of inflammatory cells (e.g., neutrophils, macrophages) in the lungs. This sustained inflammation is a hallmark of COPD, contributing to tissue damage and remodeling. Activation of NF-κB in airway epithelial cells and alveolar macrophages results in the production of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) and chemokines (e.g., CXCL8), perpetuating the inflammatory milieu. Moreover, NF-κB activation is linked to the induction of MMPs, which degrade extracellular matrix components, leading to airway remodeling and destruction of lung parenchyma [37–40].
The JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway is another crucial signaling mechanism in COPD. This pathway is activated by various cytokines and growth factors, leading to the transcriptional regulation of genes involved in cell survival, proliferation, and immune responses. In COPD, the JAK-STAT pathway modulates the expression of cytokines and chemokines that recruit and activate immune cells, exacerbating lung inflammation. For instance, IL-6 and IL-17 signaling through the JAK-STAT pathway enhances the recruitment of neutrophils and other immune cells to the lungs, contributing to chronic inflammation and tissue damage. Additionally, JAK-STAT signaling influences the behavior of structural cells in the lung, such as epithelial and smooth muscle cells, promoting processes such as apoptosis, fibrosis, and airway hyperresponsiveness [41–44].
The persistent activation of NF-κB and JAK-STAT pathways underscores their central roles in COPD pathogenesis. These pathways not only drive chronic inflammation but also mediate the destructive processes that characterize COPD, such as extracellular matrix degradation, tissue remodeling, and fibrosis. Targeting these pathways presents potential therapeutic opportunities to mitigate inflammation and prevent disease progression. For example, inhibitors of NF-κB and JAK-STAT signaling are being explored for their potential to reduce inflammation and protect against lung damage in COPD patients.
With the development of high-throughput sequencing technology, transcriptome sequencing (RNA-Seq) technology can provide technical support for the in-depth study of the molecular mechanism of COPD. RNA-Seq technology has been widely used to study the molecular pathogenesis of COPD, and to explore potential biomarkers and therapeutic targets [18]. Research has shown that COPD patients exhibit different gene expression patterns and functional pathway abnormalities compared to healthy control groups. These functional abnormalities and different expression patterns are closely related to typical characteristics of COPD, such as inflammatory response, cell apoptosis, cell metastasis, cell taxis, and oxidative stress.
In this study, potential biomarkers and therapeutic targets of COPD were explored by combining RNA-Seq sequencing technology and high-throughput expression chip technology. Effective gene markers and therapeutic targets should not only be specifically differentially expressed in tissues, but also be easily detected in blood circulation for their presence and differences. Therefore, we first screened gene expression differences at the tissue level from lung tissue samples and healthy control samples of COPD, and then, combined with gene expression information in COPD peripheral blood, we can screen genes related to the pathogenesis of COPD at different levels. The existing experimental confirmation and functional prediction of COPD related genes also provide a reliable source for us to study the molecular mechanism of COPD. By combining these differentially expressed genes and functionally related genes, we constructed a protein interaction network and analyzed the network modules to identify potential analytical markers and therapeutic targets for COPD.
A total of 850 differentially expressed genes were screened through differential gene analysis in COPD tissue, including 712 differentially expressed protein-coding genes and 138 non-coding genes. Gene Ontology (GO) function enrichment analysis found that differential protein coding genes played an important role in important cell processes such as cell chemotaxis, cell migration, and cell response to the outside world. In the process of COPD, it will cause airway and lung inflammation, which will lead to the accumulation of immune cells (including neutrophils, monocyte and lymphocytes) in the lung [37, 38]. As inflammation intensifies, the patient’s alveoli are destroyed, and some cellular components escape into surrounding tissues or circulation, which in severe cases can trigger a systemic inflammatory response. At the same time, in these complex physiological processes, some cytokine mediated signal regulation has also undergone abnormalities [39, 40], such as the intrinsic apoptotic signaling pathway, cytokine secretion, positive regulation of cell adhesion, and chemokine-mediated signaling pathway. Functional analysis of the KEGG pathway revealed that differentially expressed genes at the tissue level are involved in multiple signaling pathways associated with apoptosis, cell proliferation, cell signaling, as well as immune response. The PI3K-Akt signaling pathway [13] is crucial in regulating cell generation, differentiation, and metabolism. The NF-κB signaling pathway, which is significantly activated at the inflammatory response level, can lead to the release and oxidation of inflammatory factors and other functions [16]. In addition, important pathways such as the IL-17 signaling pathway, apoptosis, p53 signaling pathway, and TNF signaling pathway are also involved in the mechanism of COPD.
Based on known genes related to COPD, we constructed a protein interaction network. By mining the network modules, we discovered a closely interacting regulatory network module that may be closely related to the pathogenesis of COPD. The genes in the network were sorted according to their MCC importance, with the final three important genes being IL6, IL1B, and PTX3. Interleukin 6 is a cytokine with extensive biological functions in immunity, tissue regeneration, and metabolism. It is an effective inducer of inflammatory acute phase reactions, and its rapid production helps host defense during infection and tissue damage. In addition, IL-6 plays an important role in the induction of B cell and T cell differentiation [41, 42]. Interleukin-1β is an effective pro-inflammatory cytokine that plays an important role in the inflammatory response, immune stress response, and T/B cell activation, and is closely related to the pathogenic process of COPD [43, 44].
Pentraxin 3 is an acute-phase protein involved in innate immunity, inflammation regulation, and tissue remodeling. Elevated levels of PTX3 have been associated with various inflammatory conditions, including COPD. PTX3 contributes to COPD pathogenesis by: (A) Activating inflammatory pathways: PTX3 can activate Toll-like receptors (TLRs) and downstream signaling pathways, promoting the production of pro-inflammatory cytokines and chemokines, leading to chronic inflammation in the lungs [45, 46]. (B) Modulating immune responses: PTX3 regulates the activity of macrophages and other immune cells, enhancing their ability to respond to infections and tissue damage. This heightened immune activity can exacerbate lung inflammation and tissue destruction in COPD. (C) Facilitating tissue remodeling: PTX3 is involved in the regulation of extracellular matrix components and fibroblast differentiation, contributing to airway remodeling and fibrosis observed in COPD patients.
Cytochrome P450 family 1 subfamily B member 1 is an enzyme involved in the metabolism of xenobiotics and endogenous compounds, including steroid hormones and fatty acids. In COPD, CYP1B1 over-expression has been linked to: (A) Oxidative stress: CYP1B1 catalyzes reactions that generate reactive oxygen species (ROS), contributing to oxidative stress in lung tissues. Oxidative stress is a key driver of cellular damage, apoptosis, and inflammation in COPD [47, 48]. (B) DNA damage: CYP1B1-mediated oxidative stress can lead to DNA damage in alveolar and bronchial epithelial cells, resulting in cell cycle disruption and apoptosis, thereby contributing to the pathogenesis of COPD [49–51]. (C) Inflammatory responses: CYP1B1 can metabolize environmental toxins, such as polycyclic aromatic hydrocarbons (PAHs), into reactive metabolites that exacerbate inflammatory responses in the lungs, further promoting COPD progression [52, 53].
Regular monitoring of PTX3 levels in COPD patients could facilitate early detection of disease exacerbations, assessment of disease progression, and evaluation of treatment efficacy. Pentraxin 3, as an acute-phase protein, is significantly elevated during inflammation and infection, making it a useful biomarker for monitoring inflammatory status in COPD. Measurement of CYP1B1 expression levels in lung tissues or circulating cells can provide insights into the oxidative stress status of patients. Over-expression of CYP1B1 has been linked to increased oxidative stress and inflammation, both of which are critical aspects of COPD pathogenesis.
Combining PTX3 and CYP1B1 with other known COPD biomarkers, such as CRP and IL-6, could enhance diagnostic accuracy and enable personalized management strategies. CRP and IL-6 are established markers of inflammation, and their levels correlate with disease severity and progression in COPD. By integrating PTX3 and CYP1B1 measurements, clinicians could gain a more comprehensive understanding of a patient’s inflammatory and oxidative stress status, leading to more tailored and effective treatment plans.
Furthermore, PTX3 and CYP1B1 hold promise as therapeutic targets. Inhibiting the activity of CYP1B1 could potentially reduce oxidative stress and mitigate lung tissue damage. Similarly, targeting PTX3 could help modulate the inflammatory response, preventing exacerbations and slowing disease progression. The development of drugs that specifically target these molecules could revolutionize COPD treatment by addressing key aspects of the disease’s molecular pathology.
In summary, PTX3 and CYP1B1 represent potential clinical markers and therapeutic targets for COPD. Their expression profiles and functional roles in COPD pathogenesis underscore their significance in the disease’s molecular landscape. Future research should focus on validating these findings through clinical studies and exploring the therapeutic potential of targeting these molecules in COPD treatment.
Expanding on the clinical applications, the identification of PTX3 and CYP1B1 as core genes in COPD could lead to the development of novel diagnostic tools and therapeutic strategies. For instance, assays to measure PTX3 and CYP1B1 levels in blood samples could be developed for routine clinical use, allowing for regular monitoring of COPD patients. Additionally, the therapeutic modulation of these genes and their pathways could be explored through small molecules, antibodies, or gene therapy approaches.
Overall, the incorporation of PTX3 and CYP1B1 into clinical practice holds the potential to significantly improve the diagnosis, monitoring, and treatment of COPD, ultimately enhancing patient outcomes and quality of life.
The data analysis in this study is retrospective in nature, which may limit the generalizability of the findings. Prospective clinical validation is essential to confirm the potential of PTX3 and CYP1B1 as biomarkers and therapeutic targets in COPD. Future studies should include longitudinal patient cohorts to validate these findings and explore the clinical utility of these markers in real-world settings.
This study successfully integrated RNA-Seq and gene expression chip data to uncover potential molecular markers and therapeutic targets for COPD. Our comprehensive analysis identified 850 differentially expressed genes, highlighting the persistent activation of NF-κB and JAK-STAT signaling pathways as key drivers of chronic inflammation and tissue remodeling in COPD. Among the identified genes, PTX3 and CYP1B1 emerged as significant biomarkers and therapeutic targets due to their roles in inflammation, oxidative stress, and tissue remodeling.
The elevated expression of PTX3 and CYP1B1 in COPD patients suggests their potential utility in disease monitoring and therapeutic intervention. PTX3, involved in regulating immune responses and tissue remodeling, and CYP1B1, linked to oxidative stress and DNA damage, underscore their significance in COPD pathogenesis. Our findings suggest that regular monitoring of PTX3 and CYP1B1 levels, alongside established biomarkers such as CRP and IL-6, could enhance diagnostic accuracy and enable personalized management strategies for COPD patients.
Moreover, targeting PTX3 and CYP1B1 could offer novel therapeutic avenues. Inhibiting CYP1B1 activity might reduce oxidative stress and lung tissue damage, while modulating PTX3 could attenuate inflammation and prevent disease exacerbations. These insights pave the way for future research to develop diagnostic tools and therapeutic strategies focused on these molecules, potentially revolutionizing COPD treatment.
However, the study’s retrospective nature and limited sample size necessitate further validation. Prospective clinical studies with larger cohorts are essential to confirm the clinical utility of PTX3 and CYP1B1 as biomarkers and therapeutic targets. Additionally, integrating clinical testing information and other omics data will further substantiate their feasibility and effectiveness.
In conclusion, PTX3 and CYP1B1 represent promising molecular markers and therapeutic targets for COPD. Future research should focus on prospective validation and clinical translation of these findings, aiming to improve diagnosis, monitoring, and treatment of COPD, ultimately enhancing patient outcomes and quality of life.