Sepsis, a life-threatening organ dysfunction caused by a dysregulated host response to infection, affects over 19 million people annually worldwide [1–3]. It involves excessive immune activation, leading to systemic inflammatory response syndrome (SIRS) and a potential multiple organ failure [4]. Current treatments include antibiotics, fluid resuscitation, and mechanical ventilation; however, management challenges persist [5]. Neutrophils play a crucial but incompletely understood role in sepsis pathogenesis [6]. Recent advances in single-cell sequencing and Mendelian randomization have offered new opportunities for investigating disease mechanisms [7]. These technologies enable a comprehensive analysis of neutrophil function and regulatory networks at an unprecedented resolution [8]. This study integrated single-cell RNA sequencing with Mendelian randomization to elucidate neutrophil-associated molecular mechanisms in sepsis. By analyzing neutrophil transcriptomics and applying causal inference approaches, we aimed to uncover the functional alterations and regulatory networks underlying the pathogenesis of sepsis. Our findings provide novel insights into the mechanisms underlying sepsis and inform therapeutic development.
Methods
Study design
This study utilized two single-cell RNA sequencing datasets, GSE167363 and GSE95233, and applied data preprocessing and analysis workflows, including data filtering, normalization, dimensionality reduction, clustering, annotation, and pathway analysis. These methods systematically explore the mechanisms underlying the involvement of key genes in sepsis. In addition, their functionality was validated using clinical studies.
Data source
Gene expression data were obtained from the GEO database, including the GSE95233 series and GSE167363 single-cell data, involving 73 and 12 patients, respectively. The eQTL data were obtained from the eQTLGen consortium, which focuses on the genetic architecture of blood gene expression. Outcome data were sourced from the IEU database (ieu-b-69), including 10,154 sepsis cases and 454,764 controls without sepsis.
Single cell analysis
First, the expression profile was loaded using the Seurat package, and genes with low expression were filtered out (nFeature_RNA > 200 & percent.mt < 3MAD & nFeature_RNA < 3MAD & nCount_RNA < 3MAD & percent.ribo < 3MAD). Abnormal values were defined as three median absolute deviations (MAD) from the median. The data were then standardized, normalized, and analyzed using the PCA. The optimal number of principal components was determined using an Elbow Plot, and the spatial relationships between clusters were visualized using UMAP. Clusters were annotated using known cell markers, focusing on those involved in the disease onset.
Functional analysis
Functional correlations between important genes were explored using the Metascape database (www.metascape.org). Gene Ontology (GO) pathway analysis was conducted with a significance threshold of Min overlap ≥ 3 and p ≤ 0.01.
MR analysis
Outcome IDs filtered through the IEU database were extracted from the GWAS summary data (https://gwas.mrcieu.ac.uk/). Causal relationships in eQTL were selected based on a significance threshold (p < 1e-8), identifying SNPs as potential instrumental variables (IVs). SNP linkage disequilibrium (LD) was calculated, retaining SNPs with R2 < 0.001 (clumping window size of 10,000 kb) and p2 < 5e-5. Several methods, including inverse variance weighted (IVW), MR Egger, weighted median, and weighted mode, are applied to assess causality. The Wald ratio was used to evaluate the reliability of the causal relationship, estimating the impact of cis- and trans-gene expression in whole blood on sepsis.
Sensitivity analysis
We used Mendelian randomization (MR) leave-one-out sensitivity analysis to assess the impact of specific genetic variants on sepsis risk. This method systematically removes each SNP, recalculates the pooled effect size, and evaluates the robustness of the results by comparing the point estimates and 95% confidence intervals after each SNP removal. This allowed us to assess the unique contribution of each SNP and the reliability of our overall analysis.
Immune infiltration analysis
The ssGSEA (single-sample gene set enrichment analysis) method was used to assess immune cell types within the microenvironment, distinguishing 29 human immune cell phenotypes, including T cells, B cells, and NK cells. This algorithm quantifies immune cells in the expression profile, allowing inference of the relative proportions of immune infiltrates.
GSEA analysis
Patients were stratified into high- and low-expression groups based on gene expression levels, and Gene set enrichment analysis (GSEA) was used to examine the differences in signaling pathways. The background gene set included annotated sets from the Molecular Signatures Database (MSigDB). Differential expression analysis focused on significantly enriched gene sets (adjusted p-value < 0.05) based on consistency scores. GSEA is commonly used in research linking disease classification to biological significance.
GSVA analysis
Gene set variation analysis (GSVA) is a non-parametric, unsupervised method for evaluating gene set enrichment in transcriptomic data. GSVA scores gene sets, translating gene-level variations into pathway-level changes to identify the biological functions of the gene sets. In this study, gene sets from the Molecular Signatures Database (version 7.0) will be used, and the GSVA algorithm will score each set to assess potential changes in biological functions across samples.
Regulatory network analysis of important genes
This study used the R package “RcisTarget” to predict transcription factors based on motifs. The normalized enrichment score (NES) of a motif is influenced by the total number of motifs in the database. Additional annotations were inferred based on motif similarity and gene sequence. To estimate motif overrepresentation, the area under the curve (AUC) for each motif-gene set pair was calculated by comparing the recovery curve of the gene set with the ordered motifs. The NES for each motif was determined using the AUC distribution with the RcisTarget.hg19.motifDBs.cisbpOnly.500bp database.
Clinical study
Peripheral blood was collected from 20 patients with sepsis and 20 healthy controls at the Department of Burn Surgery, Huaihai Hospital, Xuzhou Medical University, between April 24, 2023, and April 24, 2024. Neutrophils were isolated using a Peripheral Blood Neutrophil Isolation Kit (Solarbio). Total RNA was extracted using TRIzol (Thermo, China), followed by reverse transcription using the FastKing RT Kit and the miRcute miRNA first-strand cDNA synthesis kit (Tiangen Biotechnology). The samples were processed for real-time PCR using 2X Universal SYBR Green Fast qPCR Mix (ABclonal) and the Light Cycler 480 system (Roche). The results were calculated using the 2–ΔΔCT method. The primers used, including the internal references, are listed in Supplementary Table SI.
Western blot
5 ml human peripheral blood was drawn, and RBCs were sedimented with 3% dextran. The leukocyte-rich plasma was layered onto an equal volume of Ficoll and centrifuged at 400×g for 30 min at 4°C. The neutrophil layer was harvested, and residual RBCs were lysed with 0.83% NH4Cl on ice for 5 min. The cells were washed twice with PBS and lysed in RIPA plus 1 mM PMSF and protease inhibitors on ice for 30 min. The mixture was centrifuged at 12000×g for 10 min at 4°C, and the supernatant was quantified using the BCA assay. The supernatant was mixed with 20 µg of 2×SDS buffer, boiled for 5 min at 95°C, and stored at –80°C. SDS-PAGE was performed, and the proteins were wet-transferred to a PVDF membrane. The membrane was blocked for 1 h in 5% non-fat milk, incubated with primary antibodies overnight at 4°C, washed 3×TBST, incubated with HRP-secondary for 1 h at RT, and developed with ECL.
Results
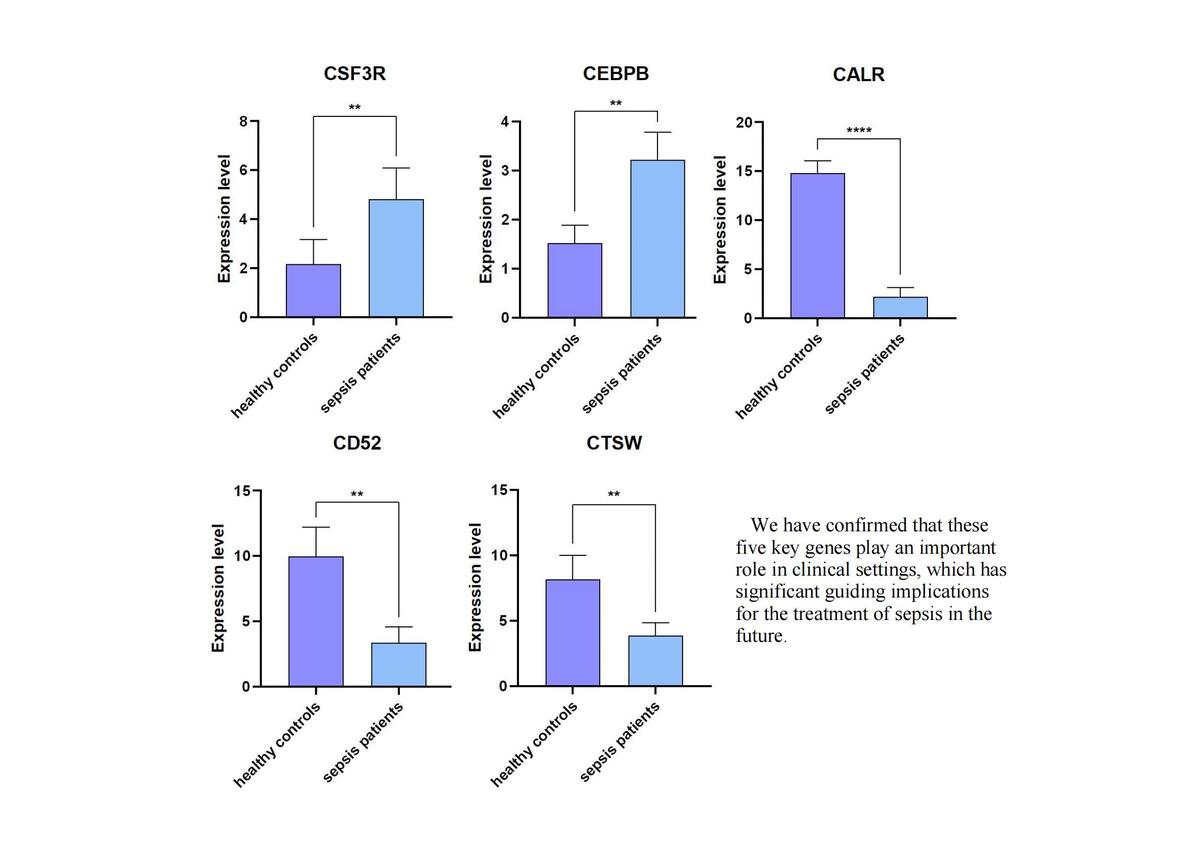
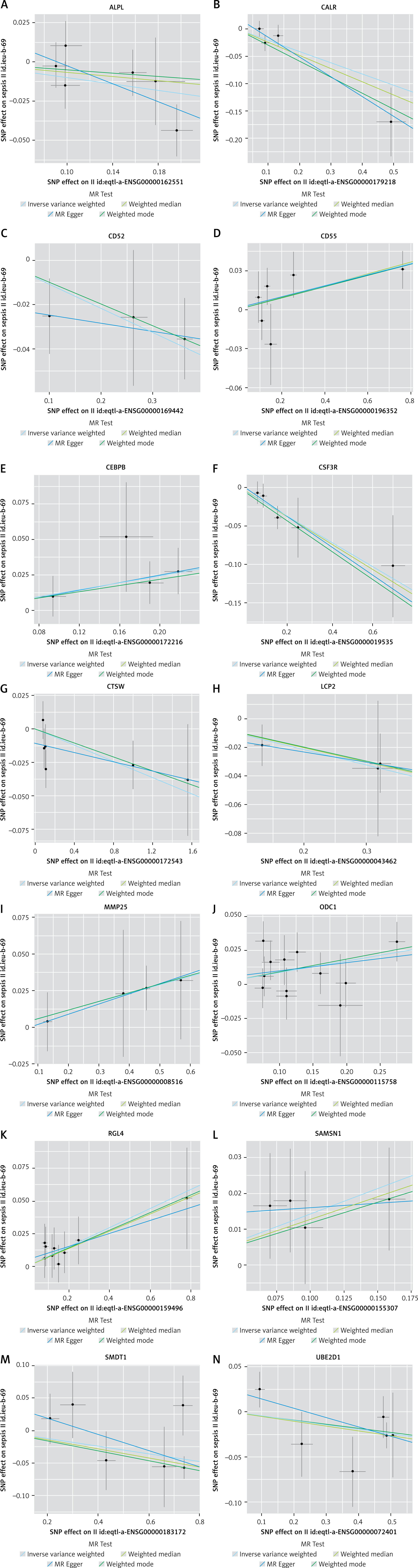
We analyzed the single-cell transcriptome data of neutrophils from the GSE167363 dataset in the GEO database (Supplementary Figures S1, S2). Mendelian randomization of differentially expressed genes led to the identification of 14 genes (Supplementary Figure S3), and degree analysis narrowed this down to five key genes (Figure 1). Using the GSE95233 dataset, ROC curves were plotted to assess the diagnostic performance of the five genes (Supplementary Figure S4). Further analysis revealed a strong correlation between these genes and immune cell infiltration in the sepsis dataset used in this study. The TISIDB database confirmed a significant relationship between the five genes and various immune factors (Supplementary Figures S5, S6). Mechanistic studies using GSEA and GSVA identified multiple signaling pathways associated with the five genes in sepsis (Supplementary Figures S7–S10). Finally, a clinical study showed altered transcription levels of these key genes in neutrophils from patients with sepsis (Supplementary Figure S11). These findings suggest that the five key genes may serve as potential prognostic indicators and therapeutic targets for treating sepsis. We extracted proteins from neutrophils for Western blot analysis, and the final results confirmed those obtained by PCR (Supplementary Figures S12, S13).
Figure 1
Mendelian randomization analysis. A–D – Scatter plots of MR analysis for important genes, with different colors representing different statistical methods. The slope of each line indicates the causal effect for each method. E–J – Scatter plots of MR analysis for important genes, with different colors representing different statistical methods. The slope of each line indicates the causal effect for each method. K–N – Scatter plots of MR analysis for important genes, with different colors representing different statistical methods. The slope of each line indicates the causal effect for each method
Discussion
Sepsis is a systemic inflammatory response syndrome caused by infection and is commonly observed in patients with severe trauma or infectious diseases [9]. It poses a major challenge in modern medicine because of the high mortality rate from complications such as organ failure and shock. The pathogenesis of sepsis is closely related to immune dysregulation and inflammation [10]. Immune cells play a critical role in sepsis by rapidly responding to pathogens through phagocytosis and cytokine secretion [11]. However, in cases of immune system compromise or highly virulent pathogens, immune responses may fail, leading to exacerbated inflammation and tissue injury.
In this study, we analyzed single-cell data from the GSE167363 dataset and identified 10 immune cell subpopulations, with seven types of immune cells: B cells, monocytes, NK cells, CD4+ T cells, platelets, neutrophils, and CD8+ T cells. We found that neutrophils, platelets, and CD8+ T cells were significantly elevated in the disease group, with neutrophils showing the greatest increase. Thus, neutrophils were selected for further investigation as they are most closely associated with sepsis.
We conducted a differential gene analysis of neutrophils and identified 468 differentially expressed genes. Using Mendelian randomization, we screened 14 genes and validated their robustness by performing a sensitivity analysis. Protein-protein interaction network analysis identified five key genes: CALR, CD52, CEBPB, CSF3R, and CTSW. CALR, CD52, and CTSW were downregulated, whereas CEBPB and CSF3R were upregulated in the high-risk group. ROC curve analysis of the GSE95233 dataset confirmed that all five genes had good diagnostic potential.
Immune infiltration is integral to sepsis, as immune cells infiltrate tissues to clear pathogens and repair damaged areas [12, 13]. However, immune infiltration can be disrupted during sepsis, leading to inadequate pathogen clearance and tissue repair, exacerbating inflammation, and contributing to organ failure. We analyzed the correlation between key genes and immune cells and found strong associations with immune factors from the TISIDB database, indicating that these genes promote immune cell infiltration and sepsis progression.
Mechanistic studies using GSEA and GSVA revealed that CALR is involved in the IL-17 signaling pathway, CEBPB in the TNF signaling pathway, and CSF3R in the reactive oxygen species pathway, all of which are relevant to sepsis. Clinical validation through qRT-PCR and Western blot analysis showed consistent results, with CALR, CD52, and CTSW downregulated and CEBPB and CSF3R upregulated in neutrophils from patients with sepsis.
In conclusion, we identified five key genes associated with sepsis and immune infiltration using single-cell and Mendelian randomization analyses. These genes have the potential to be used as diagnostic markers and therapeutic targets for sepsis.