Introduction
In the gradual progression from normal cells to cancer, these cells acquire certain acquired functions, including sustaining proliferative signaling, resisting cell death, evading growth suppressors, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis, ultimately leading to tumor formation and deterioration [1]. In recent years, as our understanding of cancer has deepened, additional characteristics of tumors have emerged, such as deregulating cellular energetics, avoiding immune destruction, genome instability and mutation, and tumor-promoting inflammation [2]. Among malignant tumors, breast cancer is the most common in women globally, accounting for 31% of all newly diagnosed cancers. It is a type of malignancy that develops through a multivariate-mediated process involving multiple steps and stages. Importantly, mutations in the BRCA1 and BRCA2 genes not only increase the hereditary nature of breast cancer but also contribute to the complexity of this disease. While early breast cancer screening and treatment advancements have led to reduced mortality rates [3], the rising incidence of breast cancer emphasizes the urgency for targeted interventions. Our study investigated lactylation in breast cancer, aiming to inform tailored treatment strategies and improve patient outcomes.
The deregulation of cellular energetics in cancerous diseases is evident in the downregulation of cell proliferation control and the adaptation of energy metabolism. Under aerobic conditions, normal cells undergo aerobic oxidation of glucose. However, in hypoxic conditions, cells further reduce the pyruvate generated from glycolysis into lactate within the cytoplasm. The Warburg effect indicates that even when oxygen is abundant, cancer cells restructure their energy metabolism by constraining the glucose metabolism process to glycolysis, leading to the production of significant amounts of lactate [4]. Glycolysis-driven energy supply is associated with cancer genes such as RAS and MYC, as well as tumor suppressor genes such as TP53. Alterations in these genes within cancer cells grant them abilities such as enhanced cell proliferation, resistance to cell death, and evasion of apoptosis, ultimately promoting tumor development [5, 6]. Lactate, a metabolic byproduct generated from glucose through glycolysis catalyzed by lactate dehydrogenase (LDH), plays crucial biological roles as an energy source, an immune regulatory molecule, and a participant in gluconeogenesis. LDH exists in two distinct subtypes, LDHA and LDHB, each with specific functions [7]. LDHA is responsible for converting pyruvate into lactate, and its expression is regulated by proteins such as hypoxia inducible factor-1α (HIF1α), c-Myc, and p53 [8]. In contrast, LDHB converts lactate back into pyruvate to promote oxidative metabolism, and its loss or downregulation is closely associated with the development and poorer prognosis of cancers such as pancreatic and liver cancer [9, 10]. Additionally, lactate produced by cancer cells can be secreted into the extracellular environment, serving as a signaling molecule to further promote cancer development [7]. It can stimulate endothelial cells to secrete VEGF protein and activate the NF-κB/IL-8 (CXCL8) pathway, thereby facilitating tumor-related angiogenesis [11, 12]. Lactate also plays a vital role in maintaining an acidic environment, regulating the tumor microenvironment (TME) through processes such as cell invasion, metastasis, and immune escape, thereby sustaining tumor growth [13]. As a result, lactate has become a potent molecule influencing the behavior of every cell within the TME.
In 2019, Zhang et al. introduced a groundbreaking concept called ‘lactylation’ – a novel post-translational modification. It involves using lactate, a product of cellular metabolism, as a small-molecule precursor to induce lactylation of histone lysine, thereby regulating gene expression. This opened up a new frontier in the study of protein lactylation. They employed mass spectrometric analysis to detect a molecular weight shift of 72.021 Daltons on histone lysine residues in the breast cancer MCF-7 cell line. Through isotopic labeling methods and various in vitro and in vivo experiments, they convincingly demonstrated the widespread presence of lysine lactylation. Furthermore, they found that the abundance of lactylation in MCF-7 cells is positively correlated with lactate concentration, and it is regulated by glycolysis and hypoxia induction [14]. Increasingly, research has shown the close association of lactylation with inflammatory diseases, tumors, neurodegenerative diseases, and more [15–17]. While the research on protein lactylation is still in its early stages, it has opened up new horizons for targeting lactate metabolism, transport, and immune-related anti-cancer strategies. Our study, based on a literature search, revealed limited reports on the functional role of lactylation in breast cancer. Therefore, our research aimed to identify differentially expressed genes related to lactylation in breast cancer, construct a prognostic model for more accurate patient prognosis prediction, and explore effective cancer therapies. Our study not only advances our understanding of the interaction between lactylation and cancer but also has the potential to uncover promising cancer immunotherapy targets, contributing to the fight against breast cancer.
Material and methods
Data download and processing
We obtained breast cancer RNA expression data, CNV files, and corresponding clinicopathological information from the TCGA-BRCA project (GDC (cancer.gov)). Clinical parameters and normalized gene expression data were obtained from the GSE162228 (GEO Accession viewer (nih.gov)) breast cancer dataset available in the GEO database, which consists of samples from Taiwanese breast cancer patients [18]. To ensure data integrity, samples lacking essential clinicopathological or survival information were excluded. Lactylation is facilitated by specific enzymes or protein modifiers. Therefore, lactylation-related genes encompass those encoding these enzymes and genes associated with the substrate proteins involved in lactylation. We included a total of 332 lactylation-related genes for subsequent analysis [19]. The lactylation-associated gene protein-protein interaction (PPI) network was constructed using the STRING website (STRING: functional protein association networks (string-db.org)). We calculated the frequency of copy number variations in lactylation-related genes by analyzing changes in gene copy numbers in breast cancer samples from the TCGA database. Subsequently, the “RCircos” package in R language was used to create a circular gene copy number map. Finally, Cox and co-expression analyses were used to generate the prognostic network of lactylation-related genes.
Screening of lactylation prognosis-related genes in breast cancer
First, we began by identifying lactylation-related genes with prognostic value through differential expression analysis and univariate Cox regression analysis within the entire dataset of breast cancer samples. Subsequently, we narrowed down the list of prognosis-related genes using LASSO regression. Genes with confirmed prognostic significance were then selected through multivariate Cox regression analysis, and we proceeded to construct prognostic models. To calculate the risk score for each breast cancer sample, we utilized the accumulation method by multiplying the coefficient with the gene’s expression level. Based on the median value, we categorized the samples into high-risk and low-risk groups and examined the prognostic differences between these groups. We employed the Kaplan-Meier method to generate survival curves for breast cancer patients, and these curves were visualized using the “survminer” package. Furthermore, we conducted an in-depth analysis of the clinical data and risk scores for all breast cancer patients, calculating survival times and statuses. This information was used to create a nomogram. Finally, we employed the R package “timeROC” (V0.4) to generate a receiver operating characteristic (ROC) curve for assessing the sensitivity and specificity of the risk model.
Cluster analysis
We employed the “ConsensusClusterPlus” package to conduct unsupervised clustering of breast cancer samples, based on the expression levels of lactylation-related genes. The results indicated that the samples were most effectively categorized into two distinct classes. Subsequently, we created a heat map to visualize the correlation between the expression patterns of lactylation-related genes in different clusters and the clinical information of patients. We then quantified the expression of immune cells in these distinct clusters using the ssGSEA method and presented the results through box plots. In addition, we obtained the GO/KEGG pathway files from the GSEA website and utilized the “GSEABase” and “GSVA” packages for pathway enrichment analysis and heat map visualization.
GO/KEGG analysis
We conducted the Wilcoxon test to identify DEGs in both groups. The risk score was calculated using the R package “limma,” with the criteria of FDR < 0.05 and |log2 FC| ≥ 1. For GO/KEGG enrichment analysis, we utilized the R packages “clusterProfiler” and “enrichplot”.
The relationship between lactylation-related molecular patterns and the clinical features and prognosis of breast cancer
To assess the clinical relevance of the clusters generated by consensus clustering, we examined their associations with molecular patterns, clinical characteristics, and survival outcomes. Clinical characteristics encompassed age, gender, tumor staging, and lymph node staging. Furthermore, Kaplan-Meier analyses were conducted using the “survival” and “survminer” packages to evaluate differences in overall survival (OS) among the various models [20].
Establishment of a predictive nomogram
The nomogram is created to offer meaningful clinical predictions for breast cancer patients, encompassing their risk scores and other clinicopathological characteristics, with a particular focus on the 1-year, 3-year, and 5-year OS rates. We assessed the clinical validity of the established nomogram through calibration curve analysis and decision curve analysis (DCA).
Lactylation-related molecular patterns and TME in breast cancer
The ESTIMATE algorithm evaluated the StromalScore and ImmuneScore of breast cancer patients, and the CIBERSORT algorithm was employed to calculate the levels of 23 immune cell subtypes for each patient [21, 22]. The infiltrating fraction of immune cells was determined using the single sample gene set enrichment analysis (ssGSEA) algorithm [23].
Drug sensitivity prediction
The half maximal inhibitory concentration (IC50) values for common anti-tumor drugs were computed using the “oncoPredict” R package to predict drug responses in breast cancer patients with varying levels of PGK1 expression.
Cell culture and transfection
The human breast cancer cell line HS578T, provided by the Medical Laboratory of Yan’an University, was utilized in this study. Cells were cultured in DMEM medium (BI, Israel) supplemented with 10% fetal bovine serum (FBS) (BI, Israel) at 37°C in a constant temperature incubator with 5% carbon dioxide. The siRNA sequence used in this research was PGK1 5′-GAGTCAATCTGCCACAGAA-3′ (Gene-Pharma, China) [24]. Previously synthesized siRNA targeting the PGK1 gene was transfected into cells using Lipo 2000 (Invitrogen, USA).
RNA isolation and quantitative real-time PCR analysis
This study utilized quantitative RT-PCR to assess the knockdown efficacy of siRNA. Total cellular RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, USA), and RNA concentration was checked. Reverse transcription was performed using Hifair III 1st Strand cDNA Synthesis SuperMix for qPCR (gDNA digester plus) from Yeasen Biotechnology, China. qPCR was conducted using Hieff qPCR SYBR Green Master Mix (No Rox) from Yeasen Biotechnology, China, with GAPDH as the reference gene. The primer sequences used in this experiment were as follows: PGK1 (forward, 5′-TCACTCGGGCTAAGCAGATT-3′; reverse, 5′-CAGTGCTCACATGGCTGACT-3′). Amplification reactions were carried out using a qPCR instrument with the following conditions: 95°C for 5 min; 95°C for 10 s; 60°C for 30 s. After 40 cycles of amplification, data analysis was conducted, ensuring correct amplification and melting curves.
CCK8 assay
In this study, cell viability of HS578T cells was assessed using the Cell Counting Kit-8 (CCK-8) method (IC-1519, InCellGene, Tx. USA). Cells were seeded at a density of 1500 cells per well in a 96-well cell culture plate and then transfected with siRNA. After transfection, cells were placed back in the incubator, and 10 µl of CCK-8 reagent was added at the same time every day for detection at 0 h, 24 h, 48 h, and 72 h. Finally, their absorbance at a wavelength of 450 nm was measured using a microplate reader (Molecular Devices, USA).
Scratch wound healing assay
HS578T cells, cultured in a 6-well plate, were transfected with both PGK1 siRNA and NC siRNA at a 70% confluence rate. A sterile 100 µl pipette tip was used to create cell scratches, and images were captured at 0, 12, 24, and 36 h post-scratch to ensure a consistent scratch area. Image acquisition was performed using a Nikon Ti-S fluorescent microscope.
Statistical analysis
All statistical analyses were conducted using R software (version 4.3.1). The t-test was employed to assess differences between the two groups, while the log-rank test was used to examine disparities between the Kaplan-Meier curves. Univariate and multivariate Cox regression analyses were performed to identify risk factors associated with breast cancer prognosis. A significance level of p < 0.05 was considered indicative of statistical significance.
Results
Lactylation-related gene expression and mutation in breast cancer
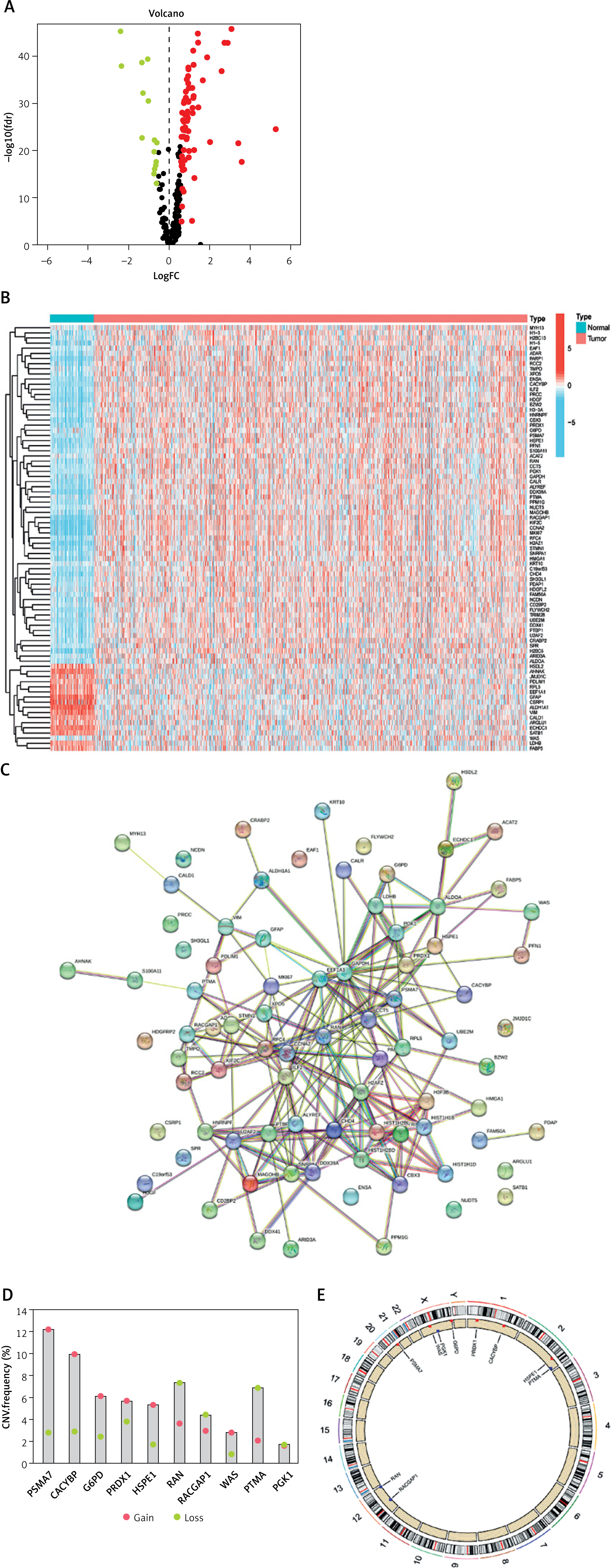
We began by identifying lactylation-related genes within the DEGs of the TCGA-BRCA dataset (Figure 1 A). Subsequently, we obtained a total of 83 DEGs for further analysis, and their relative expression levels are depicted in Figure 1 B. Using the STRING website, we conducted a PPI network analysis to elucidate interactions among these DEGs (Figure 1 C). Further, we assessed the frequencies of CNV for ten prognosis-related genes through CNV files (Figure 1 D). The results suggested that CNV potentially plays a regulatory role in the expression of lactylation prognosis-related genes. In Figure 1 E, the CNV-altered sites can be observed on the chromosomes of lactylation prognosis-related genes.
Figure 1
Lactylation-related gene expression and mutation in breast cancer. A – Difference analysis of gene expression in breast cancer. B – Differential expression of lactylation-related genes in breast cancer. C – PPI network of lactylation-related genes. D – Frequency of copy number variation of lactylation-related genes. E – Chromosomal distribution circle diagram of lactylation-related genes
Lactylation subgroups and their characterization in breast cancer
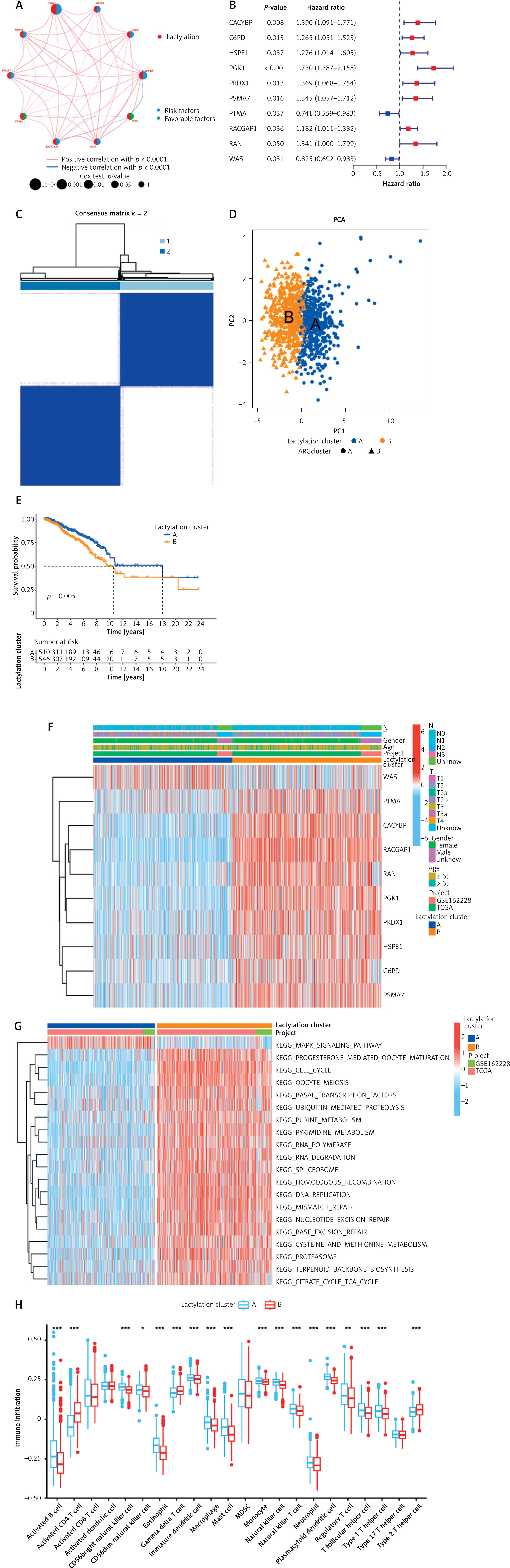
We combined the TCGA-BRCA and GSE162228 datasets to enhance the sample size and then identified ten prognosis-related genes among the 83 DEGs using univariate Cox regression analysis and Kaplan-Meier analysis (CACYBP, G6PD, HSPE1, PGK1, PRDX1, PSMA7, PTMA, RACGAP1, RAN, and WAS). Subsequently, we illustrated the interactions, regulatory relationships, and their significance for survival in breast cancer patients using a network diagram (Figure 2 A). A forest plot visually displayed the HR values of the lactylation prognosis-related genes, classifying them as high or low risk (Figure 2 B). To gain insights into the connection between lactylation and breast carcinogenesis and to determine how lactylation-related genes correlate with breast cancer expression patterns, we conducted a consensus clustering analysis of breast cancer patients based on the expression levels of DEGs. The results indicated that the optimal clustering variable was 2 (Figure 2 C), and the breast cancer patients in the cohort were well distributed into these two groups. Principal component analysis (PCA) further affirmed the clear separation between the groups (Figure 2 D). Additionally, when comparing the OS of patients in the two groups, we observed that cluster B had a worse prognosis than cluster A (Figure 2 E). Furthermore, we investigated the relationship between gene expression and clinicopathologic variables in different clusters, revealing significant differences between the two groups (Figure 2 F). We then identified differential pathways between cluster A and cluster B through GSVA analysis. These pathways included “CITRATE-CYCLE-TCA-CYCLE”, “MAPK-SIGNALING-PATHWAY”, “CELL-CYCLE”, “PURINE-METABOLISM”, “CYSTEINE-AND-METHIONINE-METABOLISM”, and “PYRIMIDINE-METABOLISM” (Figure 2 G). Lastly, we analyzed variations in immune cell infiltration levels between different clusters using the ssGSEA algorithm. The results showed that cluster B exhibited higher infiltration of activated CD4 T cells and type 2 T helper cells (Th2). Conversely, cluster A displayed more significant immune cell infiltration, including B cells, natural killer cells, eosinophils, macrophages, mast cells, monocytes, neutrophils, and other cell types (Figure 2 H). Consequently, cluster A, characterized by higher immune infiltration levels, displayed a more favorable prognosis compared to cluster B.
Figure 2
Lactylation subgroups and their characteristics in breast cancer. A – Network diagram of lactylation-related genes. B – Forest plot of lactylation- related genes at different risks. C – Unsupervised clustering of lactylation-related genes. D – PCA analysis among different clusters. E – Prognostic analysis among different clusters. F, G – GSVA analysis of signaling pathways between different clusters. H – Immune infiltration levels between different clusters
Construction and evaluation of prognostic models
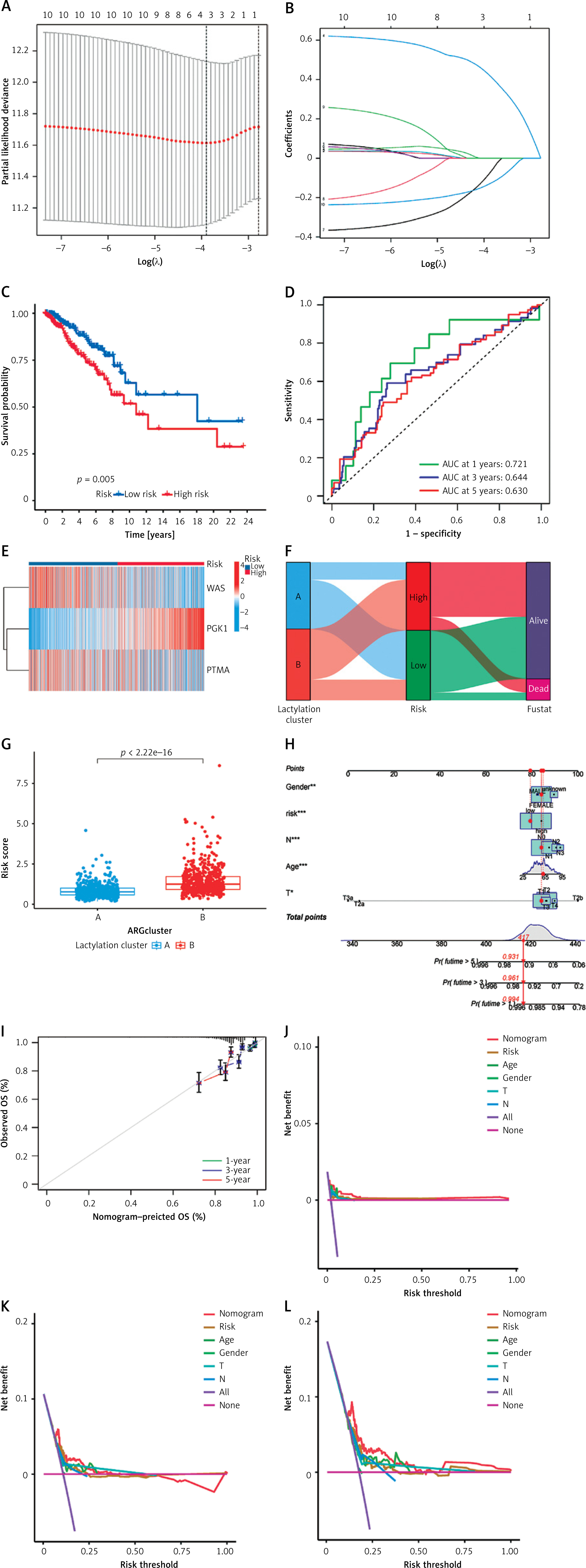
We initially identified genes associated with patient prognosis through univariate Cox regression analysis, followed by LASSO regression analysis. The LASSO analysis revealed that, based on the optimal λ-value, gene selection stabilized and minimized partial likelihood bias when including three genes (Figures 3 A, B). Consequently, we identified three lactylation-related genes significantly associated with prognosis: Risk score = (0.6409 × PGK1) – (0.3610 × PTMA) – (0.2484 × WAS). Subsequently, we divided the patients into high-risk and low-risk groups using the median risk score. Kaplan-Meier survival curves indicated that patients in the high-risk group had significantly worse prognosis than those in the low-risk group (Figure 3 C). Furthermore, we evaluated the predictive performance of this model using ROC curves. The results demonstrated high predictive accuracy, with an AUC of 0.721, 0.644, and 0.630 at 1, 3, and 5 years, respectively (Figure 3 D). Additionally, the heat map displayed the expression of the selected prognostically relevant genes (Figure 3 E). The findings suggested that WAS and PTMA might act as protective factors for breast cancer, while PGK1 could be a risk factor. We constructed a Sankey diagram to visualize the relationship between different clusters, risk scores, and patients’ survival status. These diagrams revealed that the majority of cluster A corresponded to the low-risk group with a relatively favorable prognosis, while most of cluster B corresponded to the high-risk group with a less favorable prognosis (Figure 3 F). In line with the aforementioned results, Figure 3 G indicates that the risk score of cluster B was higher than that of cluster A. Given the strong correlation between the risk score and patient prognosis, we incorporated clinical parameters to construct a nomogram. This nomogram assessed the OS of breast cancer patients at 1, 3, and 5 years (Figure 3 H). The calibration curve of the nomogram demonstrated high accuracy between actual observed and predicted values (Figure 3 I). Furthermore, the DCA curves showed that the nomogram’s prediction of patients’ OS at 1, 3, and 5 years outperformed individual clinicopathologic variables (Figures 3 J–L). Therefore, our modeled genes exhibited strong predictive efficacy, whether grouped by the risk score derived from Cox analysis or unsupervised clustering.
Figure 3
Construction and evaluation of prognostic models. A, B – LASSO regression screening of prognostic genes. C – Kaplan-Meier curves of different risk groups. D – ROC curves of different risk groups. E – Expression of modeling genes in different risk groups. F – Relationships between different clusters and risk scores, and survival status. G – Differences in risk scores between different clusters. H – Nomogram for predicting the probability of OS at 1, 3, and 5 years in breast cancer patients. I – Calibration curve for nomogram. J–L – DCA curves for nomogram
Immune infiltration analysis
As demonstrated in the preceding analysis, there is a notable disparity in patient prognosis across different risk groups. To delve deeper into the disease’s etiology and provide relevant insights for breast cancer immunotherapy, we assessed the correlation between the risk score and immune cell abundance using the CIBERSORT algorithm. The results revealed variations in the distribution and relative content of immune cells among different risk groups (Figure 4 A). Further scrutiny revealed that the risk score exhibited a positive correlation with the infiltration of macrophages M0, macrophages M2, and neutrophils, while displaying a significant negative correlation with the infiltration of naive B cells, activated CD8 T cells, and resting dendritic cells (Figures 4 B–G). Subsequently, we conducted a specific analysis of the disparities in immune infiltration levels between distinct risk groups using the ESTIMATE algorithm, which showed that the low-risk group exhibited higher immune infiltration levels (Figure 4 H). Next, we explored the relationship between genes significantly associated with lactylation prognosis and immune cell enrichment. The findings revealed a robust correlation between the two (Figure 4 I). Furthermore, we evaluated the association between the risk score and stromal cells as well as immune cells within the TME using the ESTIMATE algorithm. The results indicated that the risk score exhibited a negative correlation with StromalScore, ImmuneScore, and ESTIMATEScore, implying that the low-risk group had a higher infiltration of non-tumor cells within the TME (Figure 4 J).
Figure 4
Immune infiltration analysis. A – Distribution and relative content of immune cells in different risk groups. B–E – Correlation between risk scores and immune cell types. F–G – Correlation between risk scores and immune cell types. H – Level of immune infiltration in different risk groups. I – Correlation between modeling genes and immune cell abundance. J – Correlation between risk scores and StromalScore and ImmuneScore
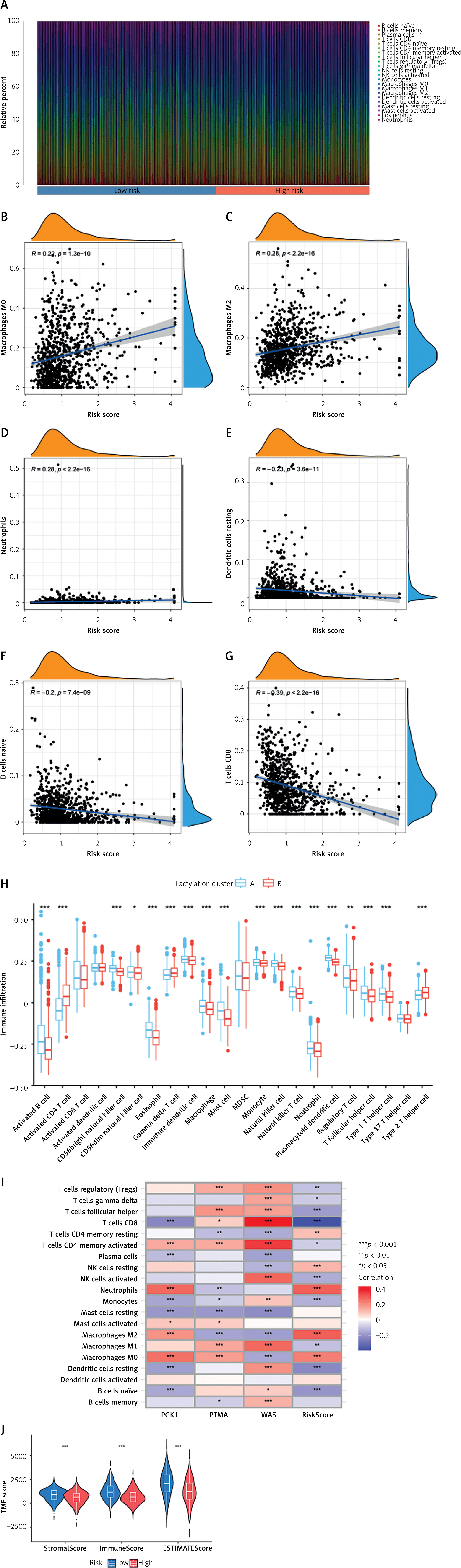
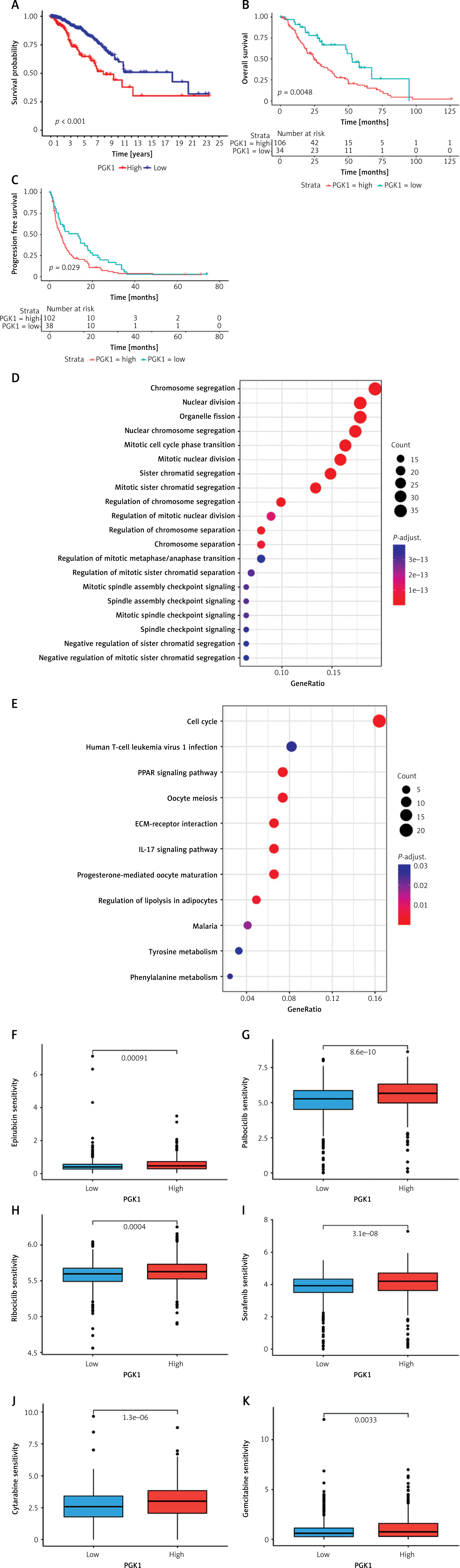
Prognostic analysis, biological function, and drug sensitivity analysis of PGK1
PGK1 exhibited the highest HR value in both Cox and LASSO regression analyses. It was also identified as one of the genes significantly associated with lactylation prognosis. Consequently, we conducted a survival analysis for PGK1. Kaplan-Meier survival curves clearly indicated that variations in PGK1 expression significantly influenced the survival outcomes of breast cancer patients (p < 0.001), with patients exhibiting low PGK1 expression demonstrating a more favorable prognosis (Figure 5 A). In the GSE124647 dataset, we also observed a significant difference in OS and progression-free survival (PFS) rates between patients with high PGK1 expression and those with low expression (Figures 5 B, C). Subsequently, we investigated the distinct signaling pathways between the high PGK1 group and the low PGK1 group through GO/KEGG enrichment analysis. Notably, we identified differentially enriched pathways such as “cell cycle”, “PPAR signaling pathway”, “IL-17 signaling pathway”, “tyrosine metabolism”, “phenylalanine metabolism”, and “ECM-receptor interaction” (Figures 5 D, E). Previous research has shown that peroxisome proliferator activated receptor (PPAR), aside from regulating energy metabolism, plays a pivotal role in immune cell differentiation and fate determination [25]; interleukin 17 (IL-17) serves as a key player in immune system regulation and is a significant pro-inflammatory factor [26]; and the extracellular matrix can impact immune function by suppressing anti-tumor immune responses [27, 28]. Hence, the signaling pathways we identified are extensively implicated in immunoregulation, energy metabolism, and cell proliferation. Lastly, we computed the IC50 values of breast cancer concerning commonly used anti-tumor drugs using the “oncoPredict” tool and compared them between the two groups. The results indicated that patients with high PGK1 expression exhibited increased sensitivity to epirubicin, palbociclib, ribociclib, sorafenib, cytarabine, and gemcitabine (Figures 5 F–K).
Figure 5
Prognostic analysis, biological function, and drug sensitivity analysis of PGK1. A – Survival curves of PGK1. B, C – Differences in OS and PFS among patients with varying levels of PGK1 expression in the GSE124647 dataset. D – GO/KEGG analysis of biological functions and signaling pathways of differentially expressed genes between the high PGK1 group and low PGK1 group. E – GO/KEGG analysis of biological functions and signaling pathways of differentially expressed genes between the high PGK1 group and low PGK1 group. F–I – Comparison of the IC50 values of common antitumor drugs between the high PGK1 group and low PGK1 group, including epirubicin, palbociclib, ribociclib, sorafenib, cytarabine, gemcitabine J–K – Comparison of the IC50 values of common antitumor drugs between the high PGK1 group and low PGK1 group, including epirubicin, palbociclib, ribociclib, sorafenib, cytarabine, gemcitabine
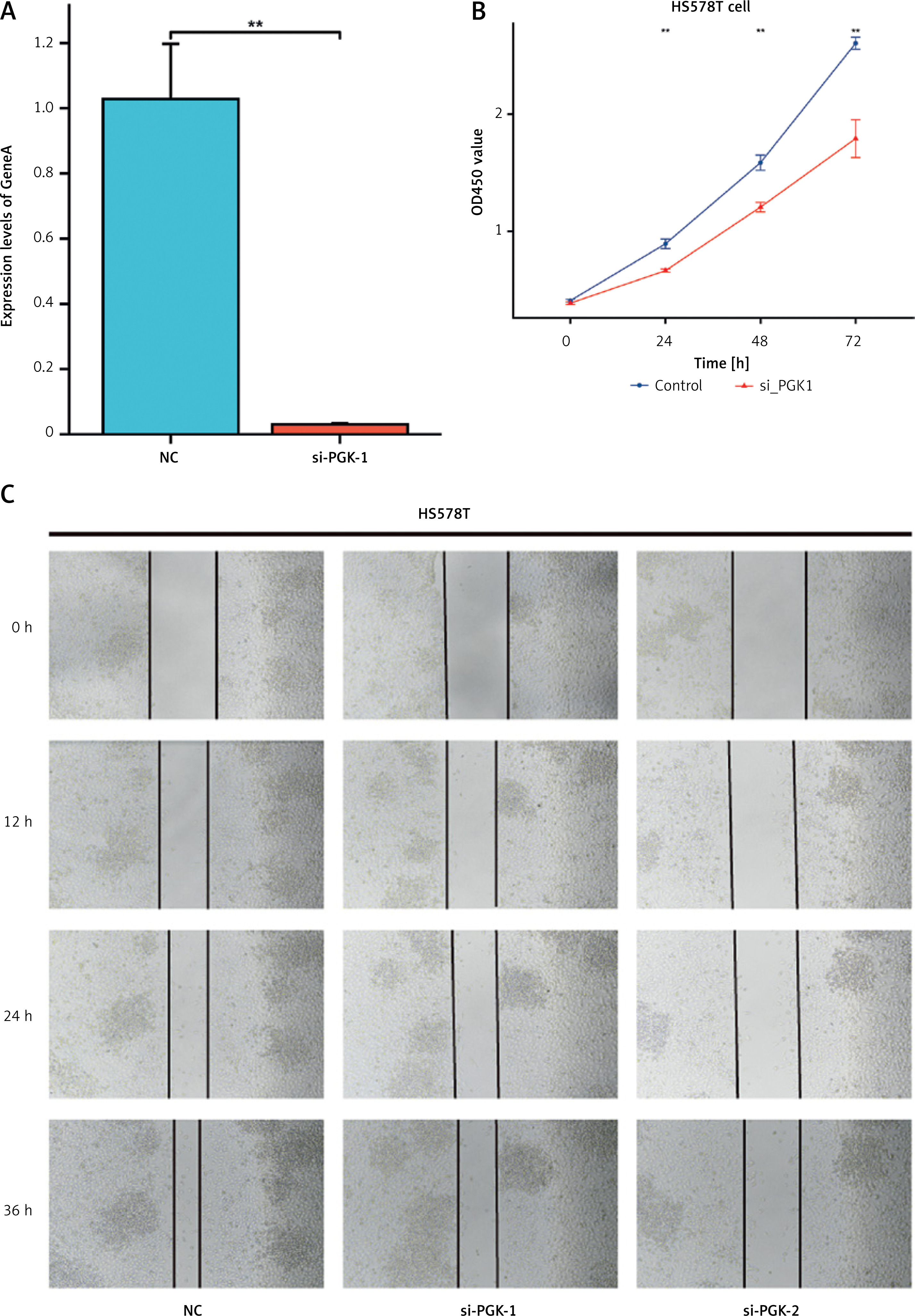
Knocking down PGK1 resulted in decreased viability of HS578T cells in vitro
We employed the quantitative RT-PCR method to assess the knockdown efficiency of PGK1 siRNA in HS578T breast cancer cells. Twenty-four hours after transfection, we examined the expression levels of PGK1 mRNA (Figure 6 A) and found a significant decrease induced by the siRNA sequences (p < 0.01). Subsequently, CCK8 analysis revealed a notable reduction in cell viability following PGK1 gene knockdown (Figure 6 B). Finally, a scratch assay was conducted to assess the impact of PGK1 knockdown on the migration capability of HS578T cells. The results indicated a significantly slower scratch closure in the PGK1 knockdown group compared to the siRNA negative control (NC) group (Figure 6 C), suggesting that PGK1 knockdown may be an effective strategy to inhibit breast cancer cell proliferation and migration.
Figure 6
Cell experiment. A – qRT-PCR assessment of PGK1 mRNA levels 24 h after transfection. siRNA sequence led to a significant decrease in PGK1 mRNA expression (p < 0.01). B – CCK8 assay. The viability of the cells was significantly reduced after PGK1 knockdown. C – Scratch wound healing assay. A significantly slower scratch closure in the PGK1 knockdown group compared to the siRNA NC group. All data are expressed as the mean ± SD of the three experimental groups. *P < 0.05, **p < 0.01, ***p < 0.001 were considered statistically significant
Discussion
Protein post-translational modification refers to the chemical alterations of proteins after translation, which can regulate protein activity, localization, folding, and interactions with other biomolecules. Proteins can undergo various forms of modification, such as acetylation, methylation, and ubiquitination. Also, with the advancement of high-sensitivity mass spectrometry, modifications stemming from cellular metabolites, such as lactylation, are gradually being discovered. In the human embryonic kidney HEK293T cell line, overexpression of histone acetyltransferase p300 (p300) has been observed to enhance lysine lactylation levels, while the absence of p300 results in a reduction in histone lysine lactylation levels in HEK293T and similar cell lines [14]. Similarly, in the lactate-induced mouse macrophage system RAW 264.7, the levels of lactylation can be significantly reduced by knocking down p300 or CREB-binding protein (CBP) [29]. Additionally, class I and class III histone deacetylases (HDACs) play a role in de-lactylation within cells [30].
The discovery of lactylation has not only opened up new frontiers in the study of protein post-translational modification but has also suggested potential regulatory mechanisms for the role of lactate in physiological and pathological processes such as cancer, inflammation, and metabolism. Lactylation levels exhibit dynamic changes in mouse oocytes and pre-implantation embryos, and in vitro hypoxic culture reduces lactylation levels, impairing the developmental potential of pre-implantation embryos [31]. Metabolic remodeling induced by GLIS family zinc finger 1 (GLIS1) involves the generation of abundant lactate and an increase in lactylation levels on pluripotency gene promoters, enhancing reprogramming efficiency and even reprogramming of aging cells [32]. Macrophage-specific expression of B-cell adapter for phosphoinositide 3-kinase (BCAP) affects the expression of repair genes by regulating lactylation levels, aiding the body in mitigating inflammatory responses [33]. Additionally, neuronal excitation in the brain elevates lactate content and lactylation levels in brain cells [34]. In Alzheimer’s disease (AD) patients’ brain samples, lactylation levels rise and become enriched at the promoters of glycolysis genes, activating their transcription. This ultimately forms a “glycolysis/histone lactylation/pyruvate kinase M2 (PKM2)” positive feedback loop, promoting the development of AD [17]. In ocular melanoma, elevated lactylation levels upregulate YTH N6-methyladenosine RNA-binding protein 2 (YTHDF2) expression, leading to the degradation of period circadian regulator 1 (PER1) and TP53 mRNA, ultimately driving tumor initiation, progression, and unfavorable outcomes [16]. Thus, exploring the role of lactylation in breast cancer becomes highly intriguing. This not only offers insights into protein post-translational modification in breast cancer research but also paves the way for new directions in the treatment of breast cancer patients.
In breast cancer, lactylation is closely associated with tumor growth, the immune microenvironment, and drug response [35]. In this study, we initially investigated breast cancer data from the GEO and TCGA databases to identify lactylation-related genes. Using an unsupervised clustering approach, we categorized breast cancer patients into clusters A and B. Among them, cluster A displayed a more favorable prognosis and higher levels of immune infiltration. Further analysis through Cox regression and LASSO regression identified three lactylation-related genes (PTMA, WAS, PGK1) that hold significant prognostic value. Among these, prothymosin alpha (PTMA) shows progressively upregulated expression in esophageal squamous cell carcinoma, with significantly higher expression levels between tumors and adjacent normal tissues as the disease progresses [36]. Circ-0004277 participates in colorectal cancer cell proliferation by upregulating PTMA expression [37]. Additionally, studies indicate that levels of PTMA in tumor samples from breast cancer patients are significantly higher than in normal breast tissue, and these PTMA levels correlate positively with certain indicators of cancer progression [38]. However, in bladder cancer, PTMA exerts its tumor-suppressive role by upregulating PTEN and coordinating the nuclear factor erythroid 2-related factor 2 (NRF2) signaling pathway through tripartite motif-containing protein 21 (TRIM21) [39]. The WAS gene belongs to the Wiskott-Aldrich syndrome protein family, and N-WASP exhibits significantly downregulated expression in breast cancer, correlating with poor prognosis [40]. Similarly, WASP acts as a tumor suppressor in T cell lymphoma [41], while in prostate cancer, it enhances cancer cell invasion and metastasis [42]. WASP and its family can also regulate actin polymerization in breast cancer, promoting cell invasion and migration, thus exhibiting oncogenic functions [43]. PGK1 is an essential enzyme in the glycolysis pathway and is involved in various biological processes. In hepatocellular carcinoma, PGK1 promotes cancer cell metastasis through pathways such as HIF-1α/PGK1 and MYC/PGK1 [44, 45]. In colon cancer, PGK1 fosters cancer metastasis by upregulating the expression of early growth response 1 (EGR1) and cysteine-rich 61 (CYR61) [46]. In papillary thyroid carcinoma, sirtuin 6 (SIRT6) enhances tumor invasiveness by increasing PGK1 expression to promote the Warburg effect [47]. Subsequently, we constructed a prognostic model using these three genes and assessed its efficacy. Patients were stratified into high-risk and low-risk groups based on the median risk score, revealing significant differences in patient outcomes between the groups. Furthermore, we analyzed the differences in immune infiltration levels between different risk groups using the CIBERSORT and ESTIMATE algorithms. The results indicated a close correlation between the low-risk group and immune cell infiltration. Following this, we developed a nomogram by incorporating the risk score and clinical-pathological parameters. The calibration curve and DCA curve both demonstrated the high accuracy of this nomogram in predicting survival rates. Given that PGK1 exhibited the highest HR in the Cox regression analysis, we explored its role further. The results revealed that patients with high PGK1 expression had significantly worse prognosis than those with low PGK1 expression. Prior research has indicated that high intracellular expression of PGK1 leads to increased tumor cell proliferation and can enhance the progression and metastasis of breast cancer through the promotion of HIF-1α-mediated EMT [48, 49]. Furthermore, PGK1’s involvement in various protein post-translational modifications such as acetylation, phosphorylation, ubiquitination, and succinylation plays a crucial role in regulating tumor metabolism and growth [50–53]. Consistent with these findings, our GO/KEGG enrichment analysis identified PGK1’s extensive involvement in immune, metabolic, and proliferative signaling pathways. Furthermore, PGK1 has been associated with chemotherapy resistance in cancer patients [54]. Finally, our drug sensitivity analysis revealed that patients with high PGK1 expression exhibited high sensitivity to anti-tumor drugs such as epirubicin and palbociclib. Other studies have also shown that inhibiting PGK1 can increase gastric cancer cell sensitivity to 5-FU and mitomycin [55], and breast cancer patients with high PGK1 expression had shorter overall survival when treated with paclitaxel [56].
Finally, we confirmed through in vitro cell experiments that the knockout of the PGK1 gene in human breast cancer HS578T cells significantly inhibits both proliferation and migration of breast cancer cells. This underscores the pivotal role of the PGK1 gene in the development of breast cancer and its potential as a promising therapeutic target for the future. Additionally, assessing PGK1 expression before chemotherapy could predict patients’ sensitivity to chemotherapy drugs, and reducing PGK1 expression presents a new strategy to overcome drug resistance. However, there is an urgent need for specific inhibitors targeting PGK1 to target cancer cells and develop therapeutic drugs, which holds significant importance. Despite some remaining questions about lactylation, the progress in related research has opened up an entirely new field in protein post-translational modification. We hope to elucidate the specific roles and regulatory mechanisms of lactylation in diseases in the near future.