Introduction
Oxidative stress has been proposed as an important parameter associated with the pathogenesis of osteoporosis, and the underlying mechanism of oxidative stress induced regulation of the Nrf2/HO-1 and NF-κB signaling pathway in osteoporosis also has been studied extensively [1, 2]. Increasing evidence reveals that reactive oxygen species (ROS) accumulation leads to oxidative stress under conditions of aging, illnesses, or medicine use, and subsequently suppresses the induction of Nrf/HO-1 and NF-κB signaling pathway activation, contributing to the genesis and progression of osteoporosis [3–5]. Therefore, antioxidants have been adopted as a promising therapeutic strategy for osteoporosis treatment.
Radix polygoni multiflori is the dried root of the polygonum plant Polygonum multiflorum Thunb. Recent studies have demonstrated that Radix polygoni multiflori exhibits antihyperlipidemic and antiatherosclerotic effects in preliminary research [6, 7]. The beneficial properties of 2,3,5,4’-tetrahydroxystilbene-2-O-β-D-glycoside (stilbene glycoside – THSG), which is regarded as the most important bioactive component of Radix polygoni multiflori, have been widely investigated, including its outstanding antioxidant and free radical-scavenging ability. Given these properties, recent studies have suggested that THSG could exert potential preventive and therapeutic effects against some chronic diseases, such as apoplexy, senile dementia (Alzheimer’s disease), hyperlipemia, and atherosclerosis [8–10].
As a transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2) is reported to mediate antioxidant related genes by binding to antioxidant response elements. Meanwhile, it has been reported that the activation of Nrf2 plays an important role in the feedback of attenuated oxidative injury via increasing activity of anti-oxidative related enzymes [11, 12]. In addition, THSG also exhibits beneficial effect on free fatty acid, superoxide anion, and focal ischemia induced tissue injury by mediating the activation of NF-κB and Nrf2 [13, 14].
In this study, the effects of THSG on oxidative damage were investigated in osteoblast-like MC3T3-E1 cells. Meanwhile, the involvement of Nrf2/HO-1 and NF-κB signaling pathways was also explored, with the aim of clarifying the potential mechanisms of THSG-mediated antioxidative protective ability in an MC3T3-E1 cell model.
Material and methods
Cell culture
The MC3T3-E1 cell line, similar to osteoblasts, was purchased from the American Type Culture Collection (ATCC). As it recommended, MC3TC-E1 was maintained in α-minimum essential medium (α-MAM) supplied with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C with 5% humidity CO2.
Cell viability assay
Methylthiazol tetrazolium (MTT) assay was applied in this study to measure cell viability. In total 1 × 105/well MC3T3-E1 cells were cultured in 96-well plates, and treated with different concentrations of THSG (0, 5, 10, 20, 50, 80, and 100 μM) for 6, 12, 24, and 48 h, respectively. THSG (originating from the root of the polygonum plant Polygonum multiflorum Thunb, molecular weight 406 and purity above 98.52%, No. 110844-200606) was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Then, cells were incubated with 0.5 mg/ml MTT at 37°C for 4 h after washing twice with PBS. Next, 200 μl of dimethyl sulfoxide was added to dissolve the produced formazan salts. An ELISA reader was used to measure the optical density at the wavelength of 490 nm, and the mean value of repeats was calculated.
Alkaline phosphatase (ALP) activity
MC3T3-E1 cells were seeded in a 12-well plate, and then cultured with α-MEM (Basal), H2O2 (100 μM), H2O2 (100 μM) + THSG (20 μM), H2O2 (100 μM) + THSG (50 μM), and H2O2 (100 μM) + THSG (100 μM) at 37°C for 4 h. After treatment, fresh medium was used to cultivate these cells for 5 days. Subsequently, cell samples were lysed using lysis buffer centrifuged at 12,000×g for 10 min. Then, supernatant was collected, and ALP activity and protein concentration in the supernatant were determined using the ALP activity assay kit (Cell Biolabs, San Diego, CA, USA) and BCA-protein assay kit (Biyuntian, Nanjing, China), respectively.
Apoptosis
MC3T3-E1 cells were cultured in a 6-well plate, and treated with α-MEM (Basal), H2O2 (100 μM), H2O2 (100 μM) + THSG (20 μM), H2O2 (100 μM) + THSG (50 μM), and H2O2 (100 μM) + THSG (100 μM) for 4 h respectively. Annexin V/PI double staining was performed to measure apoptosis according to the manufacturer’s instructions. A FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA) was used to assess the percentage of apoptosis cells.
Malondialdehyde (MDA) determination
MC3T3-E1 cells were seeded in a 6-well plate, and treated with α-MEM (Basal), H2O2 (100 μM), H2O2 (100 μM) + THSG (20 μM), H2O2 (100 μM) + THSG (50 μM), and H2O2 (100 μM) + THSG (100 μM) for 4 h respectively. The MDA concentration was determined using an MDA assay kit (Cayman, Ann Arbor, USA).
Intracellular ROS determination
MC3T3-E1 cells were seeded in a 6-well plate, and treated with α-MEM (Basal), H2O2 (100 μM), H2O2 (100 μM) + THSG (20 μM), H2O2 (100 μM) + THSG (50 μM), and H2O2 (100 μM) + THSG (100 μM) for 4 h respectively. Intracellular ROS were quantified using the ROS-sensitive dye DCFH-DA.
Real-time PCR
Total RNA was isolated using Trizol reagent (Gibco, Life Technology, Carlsbad, CA, USA) according to the manufacturer’s protocol. Reverse transcription reactions were performed as described. Then, mRNA expression was evaluated in real time on the ABI 7500 thermal cycler platform (Applied Biosystems, Foster City, CA, USA), and relative expression levels were evaluated using the 2–ΔΔCt method. Primers for each gene were as follows: 5′-TTCCTCTGCTGCCATTAGTCAGTC-3′ and 5′-GCTCTTCCATTTCCGAGTCACTG-3′ for Nrf2 (product: 215 bps); 5′-ATCGTGCTCGCATGAACACT-3′ and 5′-CCAACACTGCATTTACATGGC-3′ for HO-1 (product: 339 bps); 5′-ACTCGGAGAACTTTCAGTACC-3′ and 5′-TTGGAGCAAAGTAGAGTGGT-3′ for NQO1 3 (product: 492 bps); 5′-ATCACTGCCACCCAGAAG-3′ and 5′-TCCACGACGGACACATTG-3′ for GAPDH.
Western blot analysis
Cells were lysed in ice-cold radio immunoprecipitation assay buffer (RIPA, Beyotime, Shanghai, China) with fresh 0.01% protease inhibitor cocktail (Sigma, Shanghai, China) after treatment. Subsequently, cell lysate was centrifuged (13,000 rcf, 10 min, 4°C), and protein concentration of supernatant was determined using the BCA protein assay kit. Protein (20–30 μg) was loaded onto a 10% SDS-PAGE gel, and then transferred to a PVDF membrane (Millipore, Shanghai, China). For immunodetection, the membrane was blocked with 5% skim milk dissolved in phosphate-buffered saline with 0.05% Tween (PBST) at room temperature for 1 h. Then, blots were probed with primary antibodies against pro-caspase-3/-9, caspase-3/-9, Bcl-2, Bax, NF-κB, Nrf2, HO-1, NQO1 and GAPDH overnight, washed with PBST for 5 min and repeated 3 times. Blot membranes were incubated with goat anti-mouse or anti-rabbit secondary antibody (Beyotime, Shanghai, China). After washing with PBST, blots were visualized using the enhanced chemiluminescence method. GAPDH was used as the internal standard for western blotting.
Statistical analysis
Continuous data were presented as mean ± standard deviation (SD). Comparisons between groups were measured using Student’s t test. The difference was considered as statistically significant when p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.005). Statistical analysis was performed using SAS statistical software (SAS Inc., NC, USA).
Results
Effects of THSG on cell viability of MC3T3-E1 cells
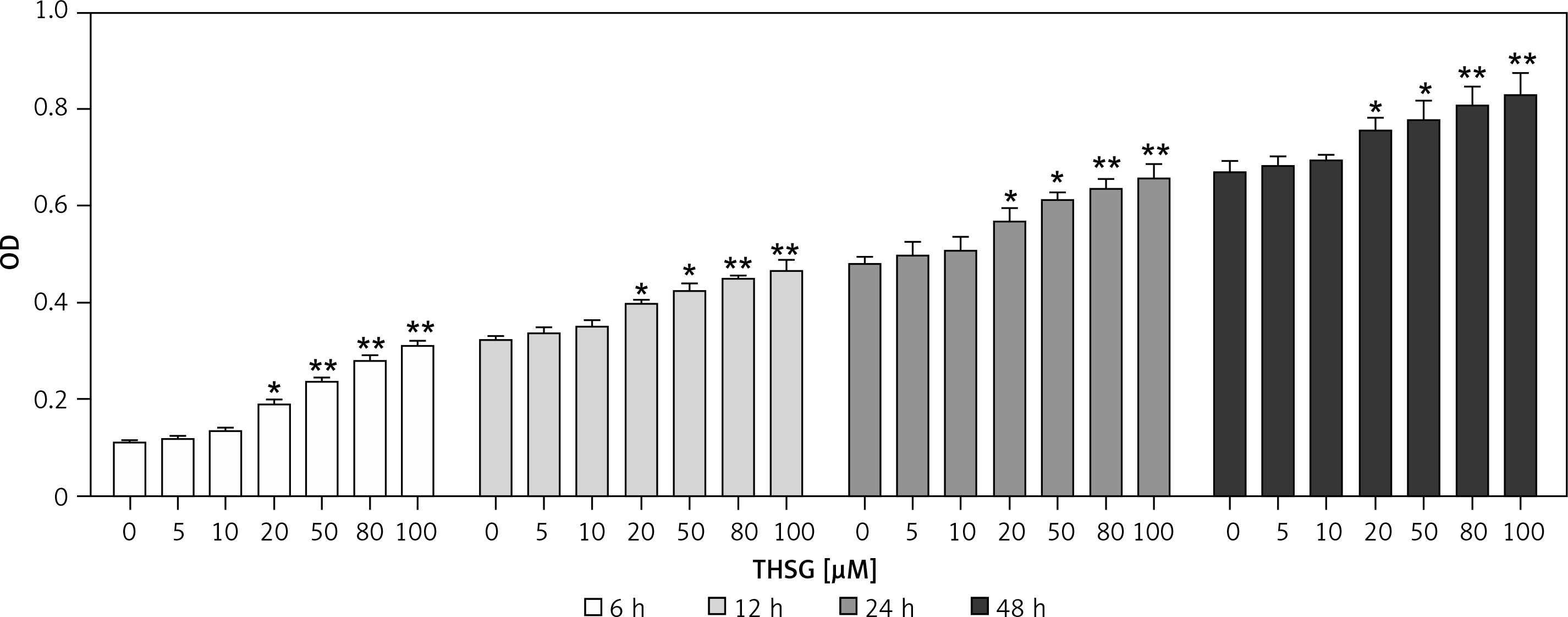
To identify the effect of THSG on proliferation of MC3T3-E1 cells, the MTT assay was carried out. As shown in Figure 1, after treatment with THSG (at 0, 5, 10, 20, 50, 80, and 100 μM) for 6, 12, 24 and 48 h, THSG over the dose of 20 μM could obviously increase the cell viability of MC3T3-E1 cells at 6, 12, 24 and 48 h in a dose- and time-dependent manner. Based on the promotive effects of THSG on cell viability, concentrations of 20, 50, and 100 μM of THSG were determined for the further investigations.
Figure 1
Effect of THSG on MC3T3-E1 cell viability. After treatment with various concentrations of THSG (0, 5, 10, 20, 50, 80 and 100 μM) for 6, 12, 24 and 48 h, MC3T3-E1 cell viability was assessed using the MTT assay as described in Material and methods (error bar = ± SD, n = 6, *p < 0.05, **p < 0.01)
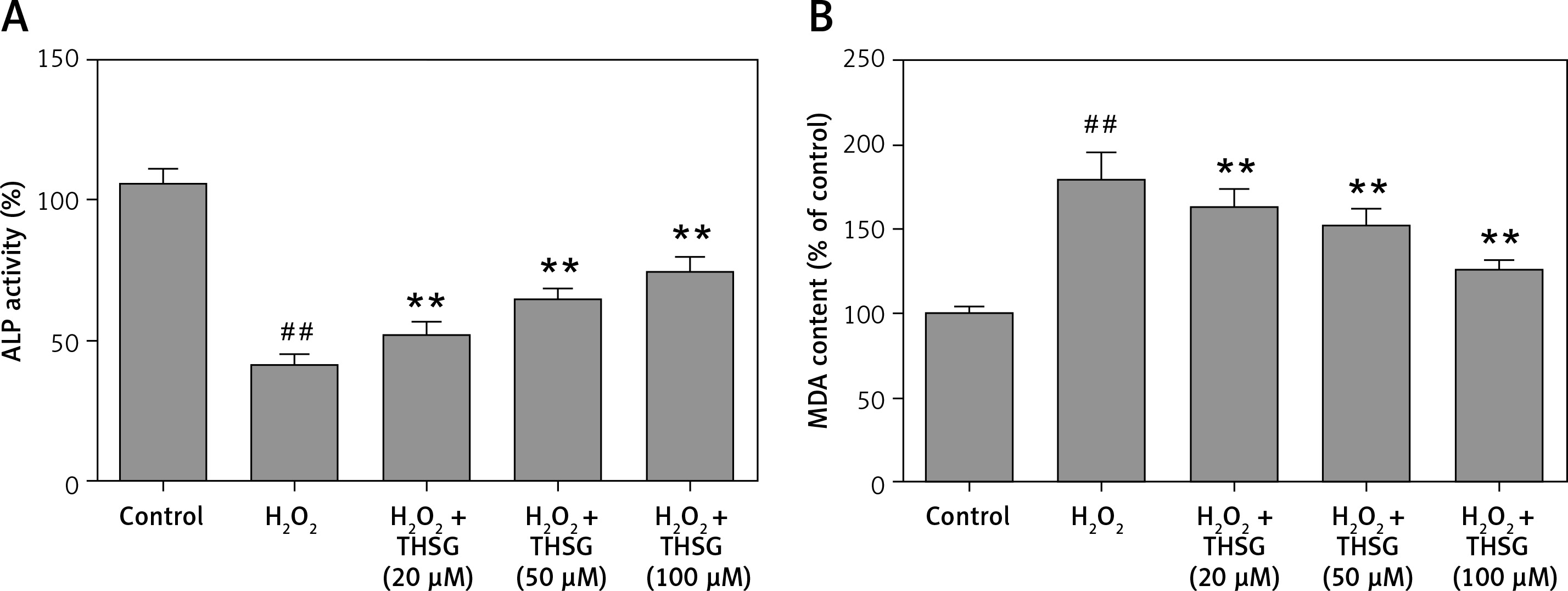
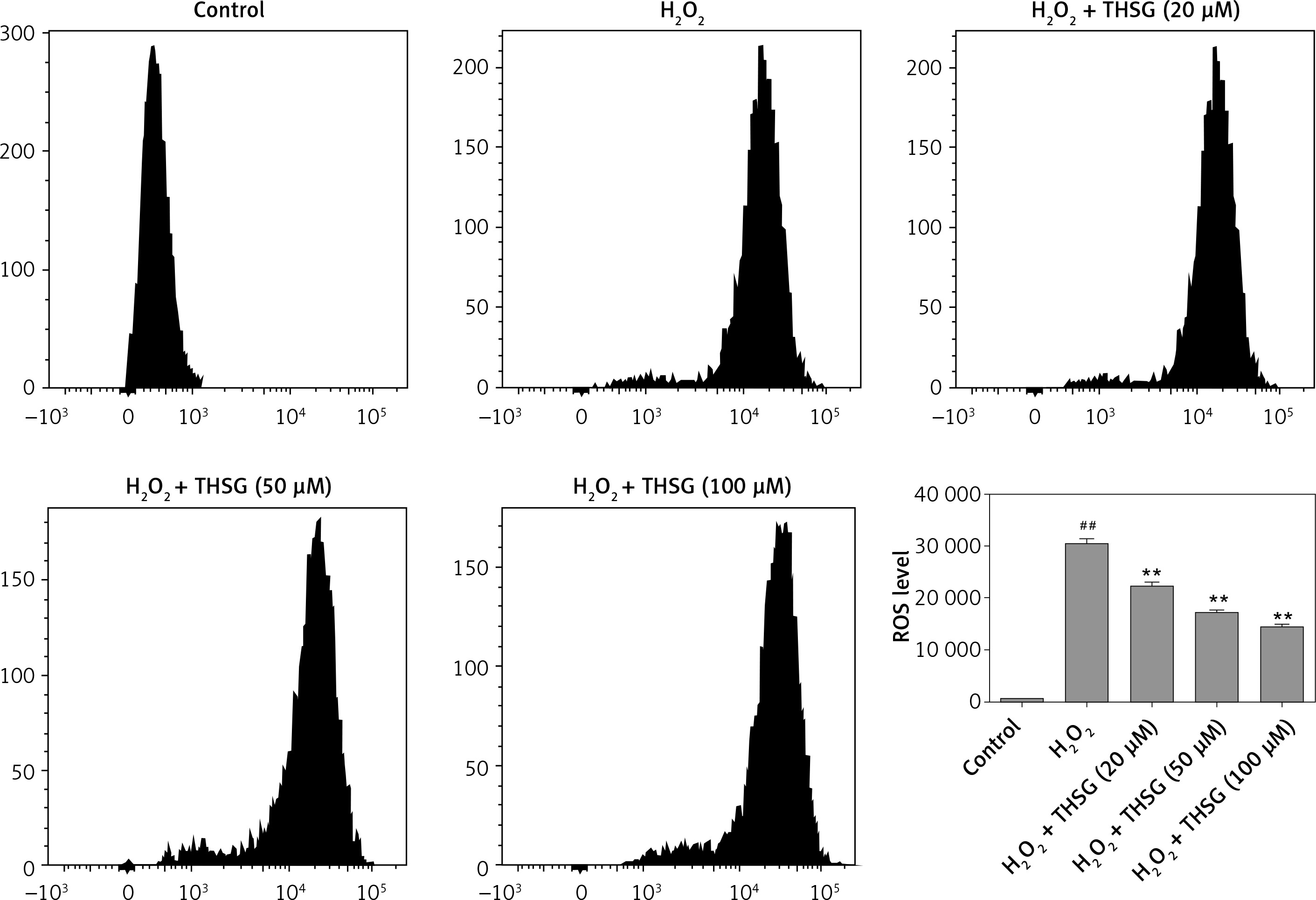
Effects of THSG on ALP activity, MDA, and ROS in H2O2-insulted MC3T3-E1 cells
Serum alkaline phosphatase (ALP) is recognized as one of the bone formation markers. In MC3T3-E1 cells, ALP activity could be significantly decreased in H2O2 treated cells compared with the controls (*p < 0.05). However, THSG significantly increased the ALP level in a dose-dependent manner (**p < 0.05, Figure 2 A). Meanwhile, the level of MDA was also assessed. The results showed that the MDA level significantly increased after treatment with H2O2 (**p < 0.05), while THSG could markedly reverse this elevation in a dose-dependent manner (**p < 0.05, Figure 2 B). These findings suggested that THSG might play an obviously negative role in the regulation of H2O2 induced oxidative damage. Thus, ROS levels in MC3T3-E1 cells with different treatments were measured. The results showed that the ROS level was significantly increased upon H2O2 treatment, but THSG intervention could markedly reduce ROS levels increased by H2O2 in a dose-dependent manner (Figure 3).
Figure 2
Effect of THSG on ALP and MDA in MC3T3-E1 cells. A – THSG attenuated decreased ALP activity induced by H2O2 treatment; B – THSG reversed the elevation of MDA induced by H2O2 treatment. In this experiment, treatments of different groups are designed as follows: control, H2O2 (100 μM), H2O2 + THSG (20 μM), H2O2 + THSG (50 μM), and H2O2 + THSG (100 μM) (error bar = ± SD, *p < 0.05, **p < 0.01, ##p < 0.01)
Effects of THSG on apoptosis of H2O2-insulted MC3T3-E1 cells
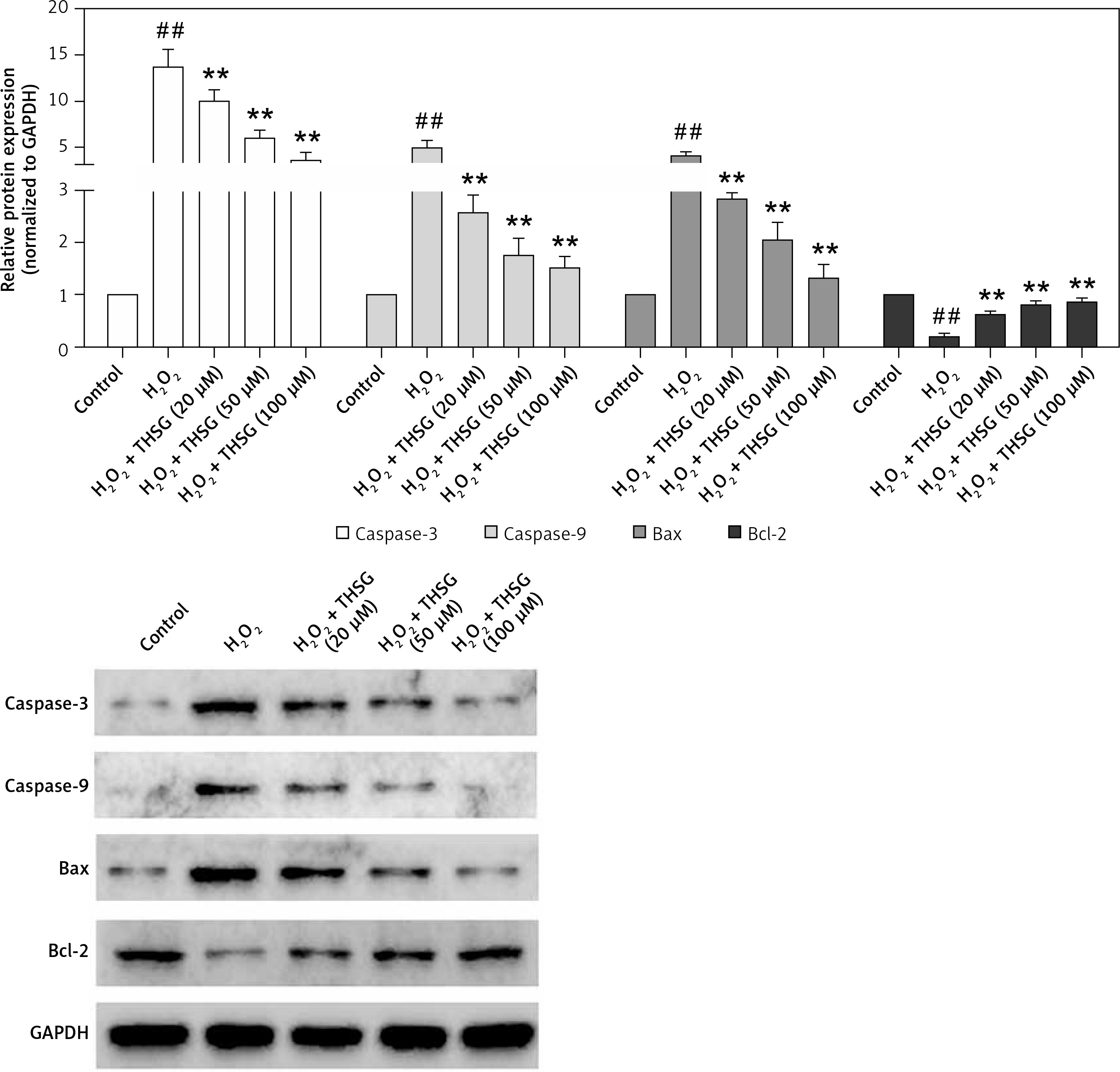
To further investigate the effect of THSG on apoptosis, H2O2 treatments were carried out using MC3T3-E1 cells. Flow cytometric results showed that H2O2 significantly promoted cell apoptosis, while THSG treatment could evidently reverse this elevation in a dose-dependent manner (Figure 4). Meanwhile, apoptosis related protein levels were also detected. The western blotting results showed that H2O2 treatment significantly up-regulated the expression levels of caspase-3, caspase-9, and Bax, and markedly down-regulated the expression of Bcl-2, while THSG could obviously reverse these effects in a dose-dependent manner (Figure 5).
Figure 4
Effect of THSG on apoptosis in H2O2 treated MC3T3-E1 cells. Cells treated with control, H2O2 (100 μM), H2O2 + THSG (20 μM), H2O2 + THSG (50 μM), and H2O2 + THSG (100 μM). Annexin V assay was used for apoptosis detection (error bar = ± SD, n = 6, *p < 0.05, **p < 0.01, ##p < 0.01)
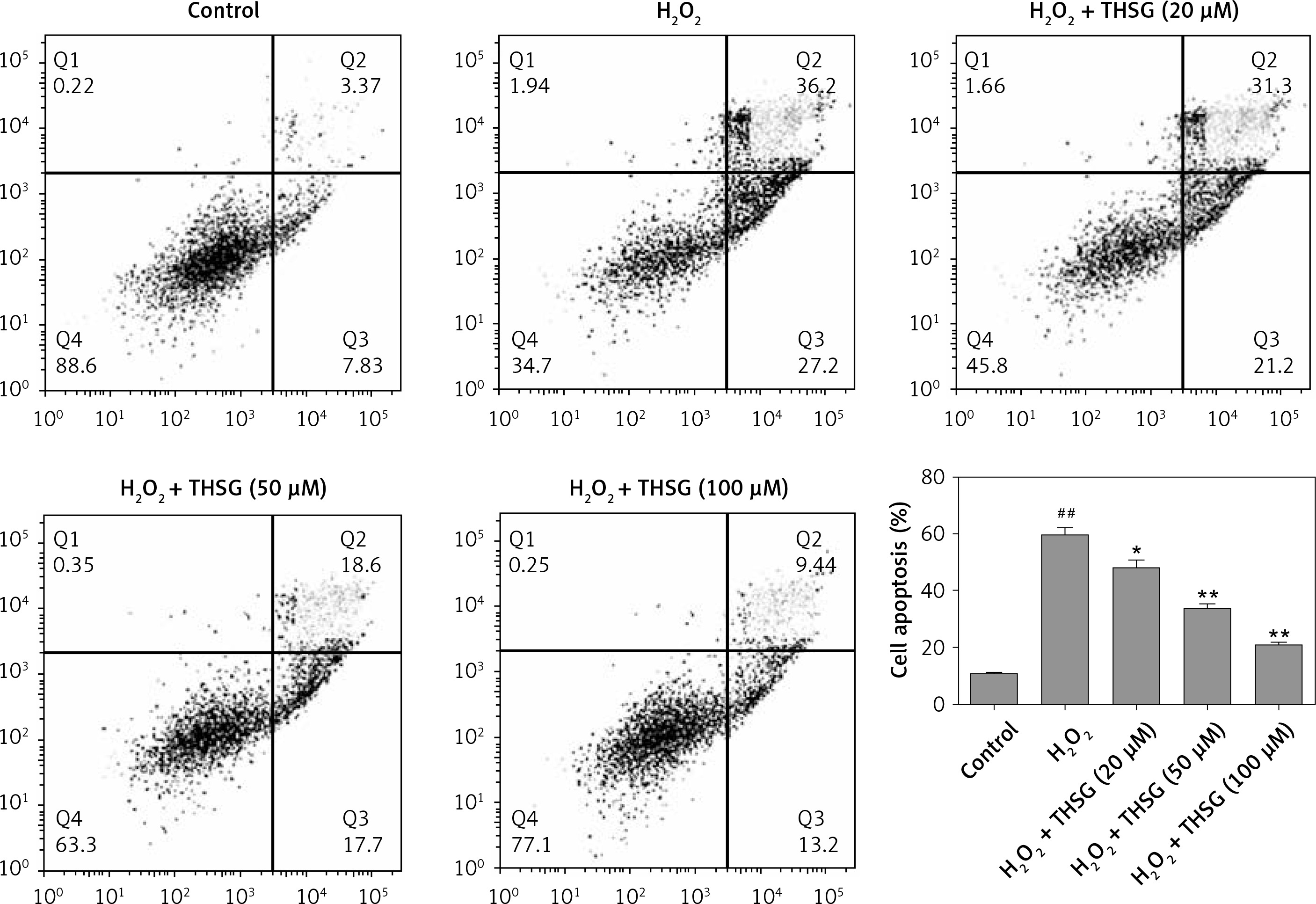
Figure 5
Effect of THSG on expression of caspase-3/-9 and Bax/Bcl-2. Cells treated with control, H2O2 (100 μM), H2O2 + THSG (20 μM), H2O2+ THSG (50 μM), and H2O2 + THSG (100 μM). Expression levels of caspase-3/-9 and Bax/Bcl-2 were determined by western blot (error bar = ± SD, n = 6, *p < 0.05, **p < 0.01, ##p < 0.01)
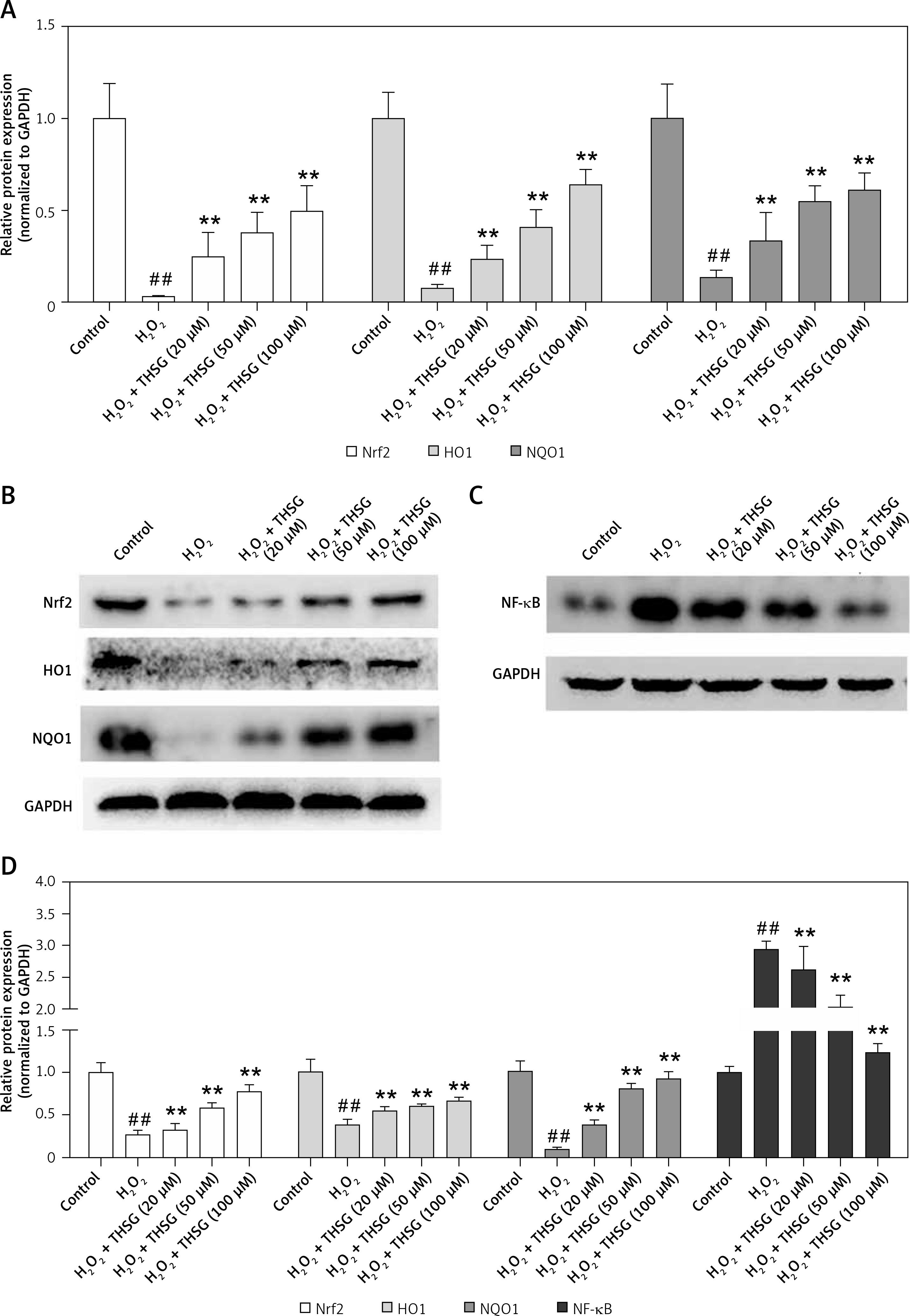
Effects of THSG on Nrf2/HO-1 and NF-κB signaling
To further reveal THSG involved signaling pathways, expression levels of Nrf2 and its downstream effectors were evaluated using RT-PCR and western blot. RT-PCR results showed that the expression levels of Nrf2, HO-1, and NQO1 were significantly decreased after treatment with H2O2, while THSG intervention could significantly reverse these elevations (Figure 6 A). Meanwhile, protein levels of Nrf2, HO-1 and NQO1 as well as NF-κB were also measured. The western blotting results showed that the protein expression levels of Nrf2, HO-1, and NQO1 were significantly lower in the H2O2 treated group than in the control group, while THSG significantly increased the expression levels of Nrf2, HO-1, and NQO1 in a dose-dependent manner (Figures 6 B, D). However, the expression level of NF-κB significantly increased in the H2O2 treated group compared to the negative control, and THSG could obviously reverse this increase, which was contrary to the variations of Nrf2 and its downstream target (Figure 6 C, D).
Figure 6
Effect of THSG on expression of Nrf2 and NF-κB pathway. A – mRNA expression levels of Nrf2, NQO1 and HO1 determined using real-time PCR. B – Protein expression of Nrf2, NQO1 and HO1 determined using western blotting. C – Protein expression of NF-κB determined using western blotting. D – Quantification of western blotting results of Nrf2, NQO1, HO1 and NF-κB. In this experiment, treatments of different groups are designed as follows: control, H2O2 (100 μM), H2O2 + THSG (20 μM), H2O2 + THSG (50 μM), and H2O2 + THSG (100 μM) (error bar = ± SD, n = 6, *p < 0.05, **p < 0.01, ##p < 0.01)
Discussion
In the present study, the bio-functional effects of THSG on oxidative damage were investigated in MC3T3-E1 cells. The results showed that THSG protected osteoblasts against oxidative damage via attenuating cell apoptosis and oxidative stress, thereby promoting cell viability. Further analysis demonstrated that the protective effect might be developed via regulating Nrf2 and NF-κB signaling pathways.
Oxidative stress acts as an important pathogenic factor for age related bone loss by regulating osteoblast and osteocyte apoptosis, osteoblast numbers, etc [15–17]. Moreover, oxidative stress has a close relationship with the pathogenesis of osteoporosis [18–20]. Statistics display a strong dependency between higher oxidative stress and lower bone mineral density in ≥ 55-year-old patients [21]. In vitro studies demonstrated that H2O2 can mediate osteoblastic differentiation, and promoted osteoblastic apoptosis. Several bone formation related metabolic markers such as ALP have been applied to investigate the effects of THSG on pathogenesis. [22]. In this study, THSG was identified to have the ability to revise the increase of apoptosis and ROS and decrease of ALP activity induced by H2O2 treatment. Mitochondria are critical for mediating the complex process of apoptosis. During this process, cytochrome c was released from mitochondria, and then interacted with caspase-9, which subsequently activate caspase-3 [23, 24] to further develop the process of apoptosis. Bcl-2 family members, including anti-apoptotic proteins and pro-apoptotic proteins, are crucial for regulating the mitochondrial death pathway [25, 26]. As an anti-apoptosis protein, Bcl-2 is located on the outer mitochondrial membrane to maintain mitochondrial permeability and to suppress cytochrome c release. In this study, expression of caspase-3, caspase-9, and Bax was significantly increased, and Bcl-2 significantly decreased after H2O2 treatment compared with the negative control, while THSG treatment could markedly reverse these changes, indicating that THSG could protect the MC3T3-E1 cells against the caspase-mitochondrial death pathway induced by oxidative stress.
The Nrf2 signaling pathway has been demonstrated as the major regulator in the meditation of endogenous and exogenous stresses induced by ROS and electrophiles [11, 27]. Thus, we explored the effects of H2O2 on the protein expression of Nrf2 and its downstream genes, including HO1 and NQO1, by RT-PCR and western blot. The results showed that Nrf2, HO1 and NQO1 were dramatically attenuated upon H2O2 treatment, and significantly up-regulated after THSG treatments. NF-κB is a negative regulator of Nrf2 expression and participates in oxidative injury induced by H2O2 [28, 29]. In this study, THSG was significantly identified to revise the decrease of Nrf2 expression and increase of NF-κB p65 in H2O2 treated cells, indicating that THSG could resist oxidative damage by regulating Nrf2 and NF-κB signaling pathways.
Collectively, THSG could attenuate oxidative injury induced by H2O2 in osteoblasts via NF-κB and Nrf2 signaling pathways. These findings might provide a possible protocol for treatment of osteoporosis, which needs an in-depth study at the molecular and cellular level to further demonstrate how THSG reduces oxidant generative diseases.