Introduction
Bladder cancer (BCa) is considered as the most common malignancy in the urinary system, with approximately 549,000 newly diagnosed cases and 200,000 deaths occuring annually [1]. In addition to certain occupational exposures to chemicals and water pollution, cigarette smoking is a major risk factor for bladder cancer [2]. There are two types of BCa: non-muscle-invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC). Among these cases, NMIBC patients accounted for about 75%, while MIBC patients accounted for about 25% [3]. Although some significant progress (surgery, chemotherapy, radiotherapy, and immunotherapy) has been achieved for BCa treatment in recent years, the 5-year survival rate was still poor and less than 60% [4]. Therefore it is warranted to clarify the molecular mechanisms and locate more effective therapeutic targets in BCa.
Collagen triple helix repeat containing 1 (CTHRC1) is a 28 KDa secreted protein that was first discovered in rat injured arteries and associated with vascular remodeling and cell migration [5]. As a positive regulator of osteoblast formation, CTHRC1 stimulates the proliferation and differentiation of osteoblasts and increases bone mass in osteoporosis [6]. CTHRC1 is linked to the proliferation of smooth muscle cells (SMC) after bladder denervation [7], and has recently been reported as a prognostic factor in multiple tumors. Kim et al. demonstrated that CTHRC1 is highly expressed and promotes cell invasiveness via activation of extracellular signal-regulated kinase (ERK)-dependent induction of MMP9 in colorectal cancer [8]. Ma et al. also identified CTHRC1 as a prognostic factor and that it facilitates cell migration and invasion through activating Wnt/PCP-Rho signaling in gastrointestinal stromal tumors [9]. According to Park et al., CTHRC1 also increases pancreatic cancer progression via activating Src, MEK and Rac1 [10]. In addition, CTHRC1 also affects the tumor aggressiveness of hepatocellular carcinoma, gastric cancer, non-small cell lung cancer, ovarian cancer, cervical cancer, oral cancer, etc. [11–16]. These reports gave rise to the hypothesis that CTHRC1 plays an important role in human cancers.
The role and molecular mechanism of CTHRC1 in BCa remains unclear. In the current study, we hypothesize that CTHRC1 acts as an oncogene of BCa development. Our data provide evidence for the first time that CTHRC1 expression is closely related to various clinicopathological features and clinical prognosis of BCa. Moreover, CTHRC1 modulates cancer progression through the PI3K/Akt signaling pathway. Our findings highlight the critical role of CTHRC1 as an oncogene and provide the basis for developing predictive biomarkers of tumor emergence and BCa therapy.
Material and methods
Patients and sample collection
Tumor tissues and matched adjacent normal tissues were collected from patients diagnosed with BCa at The First Affiliated Hospital of Zhejiang Chinese Medical University. The study was approved by the Ethics Committee of The First Affiliated Hospital of Zhejiang Chinese Medical University and written informed consent was obtained from all patients.
Cell culture and reagents
Four human BCa cell lines (EJ, 5637, J82, and T24 purchased from the American Type Culture Collection, ATCC, Manassas, VA) and a normal human uroepithelial cell line (SV-HUC-1, purchased from ATCC) were used in this study. SV-HUC-1 were cultured in F-12 medium (Gibco, Rockville, MD, USA), EJ, 5637, and T24 cells were maintained in RPMI 1640 medium (Gibco, Rockville, MD, USA) and J82 cells were cultured in DMEM medium (Gibco, Rockville, MD, USA). All media were supplemented with 10% fetal bovine serum (Sigma Aldrich, Oakville, ON) and 100 U/ml of penicillin/streptomycin (Life Technologies, Burlington, ON), and the cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
RNA isolation and quantitative real-time PCR
Total RNA from transfected cells was isolated with Trizol reagent (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed into cDNA by SuperScript II (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. qRT-PCR was performed using SYBR Prime Script RT-PCR Kits (Takara, Japan). The primers are detailed as follows: 5′-GCTAGCATGCGACCCCAGGGCCCCG-3′ (Forward) and 5′-CTCGAGTTATTTTGGTAGTTCTTCAATAAT-3′ (Reverse); β-actin, 5′-CACCATGTACCC AGGCATCGC-3′ (Forward), 5′-AGCCACCAATCCACACAGAG-3′ (Reverse). The expression of β-actin was used as an internal control by the 2–ΔΔCt method.
Cell transfection
CTHRC1 overexpressed plasmid which was ligated in the pcDNA 3.1 vector, which purchased from GenePharma (Shanghai, China). To deplete CTHRC1, two human CTHRC1-targeting siRNA sequences were synthesized and purified from GenePharma (Shanghai, China): RNAi#1: 5′-CUGGGUUGGUACUUGUUCA-3′ and RNAi#2: 5′-GCGGAGUGUACAUUUACAA-3′. Stable cell lines expressing CTHRC1-shRNA were generated by lentiviral infection. The recombinant lentivirus with the CNTN-1 gene was produced by co-transfection of 293T cells with helper plasmids (psPAX2 and pMD2G) with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the instructions.
Immunohistochemistry
Tumor sections (8 μm thick) were deparaffinized using the ethanol gradient method, followed by incubation with 3% H2O2 for 30 min to block the activity of endogenous peroxidase. The sections were then baked at 120°C for 10 min followed by staining with anti-CTHRC1 (Cell Signaling Technology, USA) and Ki67 antibodies (Beyotime, China).
Cell viability assay
The cell viability was assessed using the CCK8 proliferation detection kit (Dojindo, Tokyo, Japan). Briefly, a total of approximately 5 × 104 cells were seeded in 96-well plates and then 10 μl of CCK-8 solution was added to each well at indicated time-points (24, 48, 72 and 96 h) and cultivated for 2 h. The absorbance of the reaction system was measured spectrophotometrically at 450 nm.
Cloning formation assay
Cells were transplanted into 6-well plates and cultured at 37°C with 5% CO2 for 14 days, then colonies of cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for 30 s.
EdU labelin
Cells were incubated with 5-ethynyl-2′-deoxyuridine (RIBOBio Co, Guangzhou, China) for 3 h at 37°C, and treated with ApolloR reaction cocktail (reaction buffer and Apollo 643 fluorescence) according to the manufacturer’s protocol for 30 min, and the nuclei of the cells were stained with DAPI and visualized under a fluorescent microscope (Olympus, Japan).
Cell cycle analysis
Cells were harvested after 72 h of transfection, then digested, washed, resuspended and fixed with 75% ethanol at 4°C overnight, the supernatant was then discarded, followed by staining with propidium iodide (PI; Sigma-Aldrich; USA) and detected by using a FACScan flow cytometer (BD Biosciences, USA).
Wound-healing assay
5 × 105 cells were seeded into a 6-well plate and were cultivated until 90% confluence. A straight wound line was made using a sterile 200 μl pipette tip. Cells were cultured in the medium for an additional 24 h, and then wound recovery was evaluated under a light microscope (Olympus BX61, Japan).
Cell invasion assay
3 × 104 cells were seeded in the upper chamber precoated with Matrigel (BD, Franklin Lakes, NJ, USA) and incubated with serum-free medium. The lower chamber was filled with medium supplemented with 10% FBS. After 48 h of incubation, the invasive cells which passed through the Matrigel membrane in the lower chamber were fixed with 4% paraformaldehyde and stained with crystal violet (Cat # C-3886, Sigma-Aldrich, St. Louis, MO), then were photographed under a light microscope (Olympus BX61, Japan) at a magnification of ×100 in five random fields across the center and the periphery of the membrane.
Western blotting analysis
Cells were lysed with lysis buffer containing protease inhibitors (50 mM Tris-HCl pH 8, 50 mM NaCl, 0.5% NP-40). Equal amounts of protein were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels, and transferred to PVDF membranes. After blocking with 5% nonfat milk, the membranes were then probed with rabbit polyclonal anti-CTHRC1, anti-CDK2, anti-CDK4, anti-CDK6, anti-cyclin D1, anti-cyclin E, anti-p21cip1, anti- p27kip1, anti-E-cadherin, anti-N-cadherin, anti-vimentin, anti-p-Akt, anti-Akt, anti-p-PI3K, anti-PI3K and anti-β-actin (1 : 1000; Santa Cruz Biotechnology, CA) overnight. Following washing, the membranes were exposed to HRP-conjugated goat anti-rabbit (Abcam, USA) secondary antibodies and signals were detected with the ECL detection system.
In vivo study
All studies involving animals were performed according to the protocol approved by the Institutional Animal Care and Use Committee of Zhejiang Chinese Medical University (Zhejiang, China). Six-week old female BALB/c nude mice purchased from the animal house of Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing) were randomized to 2 groups of 6 mice each, then were subcutaneously injected into the right flank with 5 × 106 EJ (respectively transfected by sh-NC or sh-CTHRC1) cells. Tumor volumes were examined every week using the formula: volume = 0.5 × length × width2. At 28 days after the subcutaneous injection, mice were sacrificed and tumors were isolated and weighed. Mice were sacrificed after 5 weeks, and tumors were removed and weighed.
Ethics approval and consent to participate
The present study was approved by the Ethical Committee of The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang, China). Written informed consent was obtained from all enrolled patients.
Statistical analysis
All quantitative data were presented as mean ± SD and analyzed using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). The significance of differences was evaluated by a two-sided Student’s t test for two groups and one-way ANOVA test for three or more groups. The correlation between CTHRC1 expression and bladder cancer clinicopathologic characteristics was analyzed using the χ2 test or Fisher’s exact test. Survival curves were plotted with the Kaplan-Meier method and compared by the log-rank test. Survival data were evaluated using multivariate Cox regression analysis. All experiments were repeated at least three times. P < 0.05 was considered as statistically significant.
Results
CTHRC1 is elevated in human BCa tissues and cell lines
To detect the effect of CTHRC1 on BCa, qRT-PCR was used to analyze the mRNA expression of CTHRC1 in both human BCa tissues and the matched adjacent normal tissues. Significant upregulation of CTHRC1 was found in BCa tumor tissues in comparison with the matched adjacent normal tissues (p < 0.05, Figure 1 A). Similarly, western blot analysis also confirmed the upregulation of HIC1 in the BCa tissues (p < 0.05, Figure 1 B). Consistent with this result, the mRNA and protein expression of CTHRC1 was obviously high in BCa cell lines, including EJ, 5637, J82 and T24 as compared to that in SV-HUC-1 (p < 0.05, Figures 1 C, D). These data demonstrated that CTHRC1 is elevated in BCa tissues and cells.
Figure 1
CTHRC1 is elevated in human BCa tissues and cell lines. A – qRT-PCR analysis of CTHRC1 mRNA expression in BCa and matched adjacent normal tissues (n = 60). B – Western blot analysis of CTHRC1 protein expression in 5 pairs of randomly selected BCa (T) and matched adjacent normal tissues (N). C, D – qRT-PCR and Western blot analysis of CTHRC1 mRNA and protein expression in human uroepithelial cells (SV-HUC-1), and BCa cell lines (EJ, 5637, J82, and T24). E – Representative images of IHC analysis of CTHRC1 in BCa specimens (400×). F – Kaplan-Meier analysis of overall survival curves and recurrence-free survival rates in BCa patients with low and high CTHRC1 expression levels
Data are presented as mean ± SD. *P < 0.05,**p < 0.01.
CTHRC1 expression associated with clinicopathologic characteristics and poor survival in BCa patients
The clinical relevance of CTHRC1 was examined in 60 paraffin-embedded, archived clinical BCa tissues, and the IHC assay results illustrated that CTHRC1 showed stronger staining in BCa tissues (Figure 1 E). Correlation analysis of CTHRC1 expression revealed a significant relationship between CTHRC1 levels and tumor stage tumor (T) classification (p = 0.002) and node (N) classification (p = 0.013). However, CTHRC1 was not associated with age, sex, or tumor differentiation (Table I). Kaplan-Meier survival analysis suggested that patients with higher CTHRC1 expression had worse overall survival (p = 0.0023) in comparison with patients with lower CTHRC1 expression (Figure 1 F).
Table I
Correlation between CTHRC1 expression and the clinicopathological characteristics of BCa patients
Knockdown of CTHRC1 inhibits BCa cell proliferation
In light of the possible functions of CTHRC1 in BCa cells, EJ and T24 cells were transfected with si-NC, si-CTHRC1#1 and si-CTHRC1#2. Transfection efficiency was determined by qRT-PCR and western blot; the si-CTHRC1#1 sequence was selected for further study named as si-CTHRC1 for its better knock-down efficiency (Figures 2 A, B). CCK-8 and EdU assays demonstrated that CTHRC1 knockdown strikingly decreased the proliferative capacity in both cell lines (Figures 2 C, D), indicating that CTHRC1 might possess oncogene properties. Similarly, CTHRC1 silencing formed fewer colonies as compared with the sh-NC group in both EJ and T24 cells (Figure 2 E). In addition, flow cytometry revealed that CTHRC1 knockdown significantly arrests cell cycle at the G0/G1 phase in both cells, thus inhibiting the proliferation of BCa cells (Figure 2 F). Further western blot analysis was performed to evaluate protein levels including CDK2, CDK4, CDK6 (promote G1 phase progression and G1/S transition of the cell cycle), p21Cip1 and p27kip1 (block cyclin/CDK complexes and cell cycle). CTHRC1 knockdown reduced CDK2, CDK4, CDK6, cyclin D1, and cyclin E but increased p21Cip1 and p27kip1, which is consistent with the results of flow cytometry above (Figure 2 G). Taken together, knockdown of CTHRC1 inhibits cell proliferation in BCa cells.
Figure 2
Knockdown of CTHRC1 inhibits BCa cell proliferation. A, B – qRT-PCR and western blot were performed to measure the efficacy of CTHRC1 siRNA in EJ and T24 cells. C – Cell proliferation was measured by the CCK-8 and EdU assay after EJ and T24 cells were transfected with sh-CTHRC1 or sh-NC. D – Cell proliferation of EJ and T24 cells transfected with sh-CTHRC1 or sh-NC was detected by EdU incorporation assay. E – Cell colonies of EJ and T24 cells transfected with sh-CTHRC1 or sh-NC were determined using cell clone formation assay. F – Cell cycle distribution of EJ and T24 cells transfected with sh-CTHRC1 or sh-NC was detected using flow cytometric assay G – Western blot was used to examine the protein levels of proliferation markers
Data are presented at mean ± SD. *P < 0.05, **p < 0.01.
Knockdown of CTHRC1 inhibits migration, invasion and EMT of BCa cells
To elucidate CTHRC1 expression association with lymph node metastasis, wound healing assay (Figure 3 A) and transwell assay (Figure 3 B) were applied to explore the effect of CTHRC1 on BCa cell migration and invasion. The results implied that sh-CTHRC1-transduced EJ and T24 cells exhibited decreased migratory and invasive abilities. As EMT plays an important role in cell migration and invasion which is embodied in changes of certain proteins such as E-cadherin (known as epithelial markers), N-cadherin and vimentin (considered as mesenchymal markers) [17], we explored whether CTHRC1 mediated EMT in BCa cells by western blot assay (Figure 3 C). As expected, knockdown of CTHRC1 significantly increased the expression levels of E-cadherin while reducing the expression levels of N-cadherin and vimentin, signifying that CTHRC1 knockdown can reverse the EMT process of BCa cells. Collectively, CTHRC1 knockdown alleviates migration, invasion and EMT in BCa cells.
Figure 3
Knockdown of CTHRC1 inhibits migration, invasion and EMT of BCa cells. A, B – Wound healing (A) and transwell (B) assays were used to determine the cell migration and invasion of EJ and T24 cells transfected with sh-CTHRC1 or sh-NC. C, D – Western blot was used to detect the protein levels of EMT markers
Data are presented at mean ± SD. *P < 0.05, **p < 0.01.
CTHRC1 modulates the PI3K/Akt signaling pathway in BCa cells
Mounting evidence has proved that the PI3K/Akt signaling pathway plays a critical role in tumor growth and metastasis [18, 19], which encouraged us to verify whether CTHRC1 participates in modulation of the PI3K/Akt signaling pathway in BCa. Ectopic expression of CTHRC1 in both EJ and T24 cells was established by transfection of the pcDNA3.1-CTHRC1 vector. Then we examined the effects of CTHRC1 overexpression and PI3K inhibitor (LY294002) on the PI3K/Akt pathway by western blot analysis in BCa cells. The results demonstrated that CTHRC1 markedly increased the phosphorylation of PI3K and Akt in both EJ and T24 cells in comparison with the control cells (Figures 4 A, B). At the same time, the results of detection of proliferation and EMT protein markers presented the opposite effect on BCa cells compared with CTHRC1 silencing, which proved that CTHRC1 could promote proliferation, migration, invasion and EMT once again. Sure enough, overexpression of CTHRC1 combined with LY294002 () reversed activation of the PI3K signaling pathway and abolished the promotion of proliferative, migratory and invasive abilities and EMT by CTHRC1. The above data indicated that CTHRC1 promotes cell proliferation and invasion by regulating the PI3K signaling pathway.
Figure 4
CTHRC1 modulates the PI3K/Akt signaling pathway in BCa cells. A, B – The protein levels of p-PI3K, PI3K, p-Akt and Akt, proliferation and EMT markers in EJ and T24 cells were assessed by western blot analysis. The concentration of LY294002 was 5 μM
Data are presented at mean ± SD. *P < 0.05, **p < 0.01.
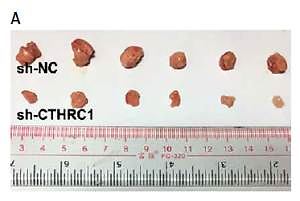
Knockdown of CTHRC1 inhibits tumorigenicity of BCa in vivo
We further explored the effect of CTHRC1 knockdown on the tumorigenicity of BCa in vivo. The results revealed that the growth of sh-CTHRC1-transfected EJ cells was significantly slower in comparison with the sh-NC group (Figures 5 A, B). Furthermore, the average weight of tumors generated with sh-CTHRC1-transfected EJ cells was largely decreased in comparison with the sh-NC group (Figure 5 C). Immunohistochemical staining revealed lower expression of CTHRC1 and Ki67 in the sh-CTHRC1 group than in the sh-NC group (Figure 5 D). These data indicate that knockdown of CTHRC1 suppresses tumorigenicity of BCa cells in vivo.
Figure 5
Knockdown of CTHRC1 inhibits tumorigenicity of BCa in vivo. A – Representative images of tumor burden in xenograft mice. B – Tumor volumes were monitored on indicated days. C – Tumor weights in each group were measured after mice were sacrificed. D – The expression of CTHRC1 and Ki67 was analyzed by IHC analysis in indicated tissues
Data are presented at mean ± SD.*P < 0.05, **p < 0.01.
Discussion
Patients with metastatic BCa are often characterized by a high rate of recurrence, progression, mortality and poor prognosis, and the potential molecular mechanisms of BCa progression and metastasis are still unclarified. Hence, early detection of the molecules involved in tumor growth, metastasis and invasion can significantly improve the incidence and clinical prognosis of patients with BCa. Herein, we first provide insights into the role of CTHRC1 as an oncogene in BCa to promote tumorigenesis and mobility.
CTHRC1 was as originally found as an extracellular matrix glycoprotein during the arterial injury-repair process [5], which has been shown to modulate vascular remodeling, osteoblastic bone formation and cell migration [6, 20]. In addition, CTHRC1 serves as a prognostic factor involved in driving human tumor progression [21–25]. In accordance with this evidence, we observed that CTHRC1 mRNA and protein were dramatically upregulated in both clinical samples and BCa cells. CTHRC1 expression was increased in bladder cancer tissues and cell lines compared with normal controls. Further clinical analysis also suggested that high expression of CTHRC1 is positively correlated with poorer overall survival and recurrence-free survival in patients with BCa, indicating that CTHRC1 is associated with BCa development, and associated with advanced clinical stage and lymph node metastasis. Also, high levels of CTHRC1 expression in patients predicted a poor prognosis. As for its biological roles in human tumors, numerous studies have manifested that CTHRC1 promotes tumor growth, invasiveness and the EMT process in colorectal cancer, gstrointestinal stromal cancer, epithelial ovarian cancer, etc. [8, 9, 13, 26]. The most common causes of BCa cancer-associated mortality are the occurrence of local invasion or distant metastasis, rather than the presence of the primary tumors themselves. In view of the most common causes of BCa-related death being the occurrence of local invasion or distant metastasis and the role of CTHRC1 in other tumors, it is speculated that CTHRC1 can regulate proliferation, invasion and EMT in BCa. As expected, our present study implied that CTHRC1 silencing dramatically inhibited BCa cell proliferation, migration, invasion and EMT in vitro and suppressed the growth of xenograft tumors in vivo.
Evidence suggested that lncRNA such as CTHRC1 might regulate several signaling pathways through microRNA [27–30]. Ma et al. reported that CTHRC1 facilitated the Wnt/PCP-Rho-JNK pathway by forming the Wnt-receptor complex [9]. CTHRC1 was also reported to activate the MEK-ERK pathway [8], JNK1/2 pathway [31], TGF-β/Smad pathway and YAP nucleus translocation [31]. Typically, it has been reported that the Akt signaling cascade and its up- and down-stream molecules including EGFR, ERK1, PI3K, and MMP, etc. can also be modulated by CTHRC1 [24, 32]. PI3K/Akt signaling has been shown to have a pivotal role in the proliferation, migration, invasion and EMT in the malignant progression of multiple tumors [33], and the importance of PI3K/Akt signaling in BCa development has been implicated [34]. Our data have illuminated an oncogenic role of CTHRC1 in BCa progression, and further study of the mechanism elucidated the biological relationship of CTHRC1 and PI3K/Akt signaling in BCa. Notably, overexpression of CTHRC1 promoted the phosphorylation of PI3K and Akt in BCa cells, which represented activation of the PI3K/Akt signaling cascade, while concomitant treatment with the PI3K inhibitor LY294002 suppressed the promoting effect by CTHRC1, confirming that CTHRC1 can induce tumor growth and metastasis in BCa through regulation of the PI3K/Akt signaling pathway.
In conclusion, our findings provided direct evidence that CTHRC1 plays an oncogenic role in BCa progression through mediating the PI3K/Akt pathway. Therefore, CTHRC1 might serve as a biomarker for early detection and prognosis of BCa, and provided a potential attractive therapeutic target for BCa treatment.