Introduction
Cervical cancer affects the female reproductive system and is the fourth most common cancer affecting women globally; approximately 528,000 new cases are identified every year. In Saudi Arabia, cervical cancer is the 13th most frequent malignancy in women, while its incidence is considerably low [1]. The early and advanced stages of cervical cancer can be treated by surgery, radiation therapy and chemotherapy. However, conventional methods have a number of disadvantages, and some cancer cells develop resistance to chemotherapy [2]. Currently used Food and Drug Authority (FDA) approved cancer drugs are generally categorized as apoptosis inducers by triggering specific pathways [3]. Apart from apoptosis, different programmed cell death (autophagy and necroptosis) mechanisms are also involved in human physiological processes [4–7]. These non-apoptotic mediated cell death mechanisms (NACD) are also specifically considered cell physiological mechanisms in which irreparably damaged DNA and/or intracellular organelles are scavenged through the phagocytosis/ubiquitin system without any other irrelevant inflammatory response [8]. Earlier studies suggested that there are a number of interconnected pathways associated with different modes of NACD in human cells. Non-apoptotic mediated cell death pathways are associated with a number of genes that are major regulators in the molecular machinery of autophagy modulation by both extracellular and intracellular signals of tumor pathophysiology [9, 10]. Specifically, an intracellular catabolic process of cells eliminates their damaged proteins and organelles through lysosomes [9, 10]. Non-apoptotic cell death-induced substances have the potential to inhibit apoptotic cell death-resistant tumor cells; thus, induction of NACD can be useful to treat cancer [11].
Autophagy, which is a non-apoptotic cell death and considered a programmed cell death process, involves the intracellular degradation of cytoplasmic components such as aged proteins, misfolded/defective proteins or ruined organelles in the lysosome [5]. Dysregulation of the autophagic pathway is linked with chromosomal instability and DNA damage, which leads to carcinogenesis [12]. The autophagy pathway is activated by various extra- or intracellular stresses, including nutrient starvation, DNA damage, reactive oxygen stress, hypoxia, mitochondrial damage, ER stress and pathogens [13]. Specifically, microtubule-associated protein 1 light chain 3 (MAP-LC3) is an autophagosomal marker that forms a preautophagosome by conjugating with an autophagosomal precursor during autophagy initiation. Finally, the cytosolic compartment is engulfed by autophagosomes by formation of a cup-shaped membrane that is then closed [14]. Interestingly, modulation of autophagy may be a target for cancer therapy and can be an alternative mode of cell death in apoptosis-resistant, cellular senescence and aging of cells [15]. Several conventional cytotoxic drugs act as autophagy inducers that show suppressed tumor growth and triggered cell death in both in vitro and in vivo models [16].
Naphthoquinones are quinone derivatives that are widely distributed in plants and exhibit excellent biological properties, including pro-inflammatory, anti-inflammatory, anti-bacterial and anti-cancer properties [17–20]. Plumbagin (PL) is a 5-hydroxy-2-methyl-1,4-naphthoquinone that has a variety of pharmacological properties such as anti-cancer, anti-microbial, anti-fertility and anti-oxidative properties [21]. The cytotoxic properties of PL and its derivatives have been studied in cancer cell lines [21–25]. Plumbagin has activated cancer cell death and cell survival-regulated signal transduction pathways [22–25]. Mostly, the PL induced cell death through ROS-mediated apoptosis by morphological alterations and DNA damage, initiating a death signal via mitochondria and activation of caspases in several cell lines [22–24]. The PL modulated p65 and Ik-Bα kinase activation that led to suppression of NF-κB activation and down-regulation of NF-κB-regulated anti-apoptotic gene expression [26]. Kuo et al. suggested that PL induced G2/M phase cell cycle arrest and inhibited the phosphatidylinositol 3-kinase/AKT pathway, leading to autophagic cell death in breast cancer cells [27]. Thus, in the present study we aimed to evaluate the effect of PL on SiHa cells to infer the specific mode of cell death as to whether cells die through apoptosis and/or non-apoptosis, providing additional confirmation supporting PL as an alternative for cervical cancer treatment.
Material and methods
Cell culture
Cancer cells (Cervical, SiHa) were obtained from the National Centre for Cell Science (NCCS), Pune, India. The cervical cancer cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin in a CO2 incubator with 5% CO2 at 37°C. Plumbagin was acquired from Sigma-Aldrich, USA.
MTT assay
The effect of PL on cervical cancer cell viability was investigated by MTT assay according to the Mossmann (1983) method [28]. About 10,000 SiHa cells per well were seeded in 96-well plates and incubated for 24 h. After 24 h, the cells were incubated with different doses of PL ranging from 1 µM to 20 µM or without PL (control) for 12 and 24 h (quadruplicates for each concentration). For the test, the medium was refreshed and 20 µl of MTT reagent [3-(4,5-dimethyl thiazol-2yl)-2,5-diphenyl tetrazolium bromide] was added at the end of the harvesting period. The plate was incubated for 4 h in the dark. The purple formazan crystals were solubilized with 100 µl of dimethyl sulfoxide (DMSO). The color developed was measured by a multiwall plate reader (Bio Rad, USA) at 570 nm with reference wavelength at 630 nm. The inhibitory concentrations (IC25, IC50 and IC75) were calculated using Calusyn software.
Acridine orange/ethidium bromide staining
Nuclear morphological changes were assessed using acridine orange (AO) and ethidium bromide (EB) staining according to Spector et al. [29]. The dye solution (AO and EB solution, one portion of 100 µg/ml AO and one portion of 100 µg/ml EB in PBS) was applied to a cover slip of cultured cells in 6-well plates, and the slides were kept in the dark at 37°C for 5 to 10 min and then examined under a fluorescence microscope (Carl Zeiss, Germany) using a 450–490-nm range UV filter.
Alkaline single-cell gel electrophoresis (comet) assay
DNA damage was analyzed using a comet assay according to Gopalakrishna and Khar [30]. Low-melting-temperature agarose in PBS was mixed with PL-treated and control cells. The mixture was pipetted out and pre-coated as a layer on microscopic slides at the normal gelling temperature of agarose. The microscopic slide layer was solidified by chilling for 10 min. Subsequently, slides were immersed in lysis buffer (100 mM Na2EDTA, Triton X-100, 2.5 M NaCl, 0.2 mM NaOH, pH 10.01 and 10 mM Tris) and incubated at 4°C overnight to allow cell lysis and DNA unfolding. After incubation, the slides were treated with alkaline buffer (1 mM Na2EDTA, 300 mM NaOH, pH > 13) for 20 min to unwind the DNA. Subsequently, slides were washed with buffer (0.4 M Tris, pH 7.5) to neutralize the alkali and remove detergents. Then, slides were stained with ethidium bromide (20 µl in 50 µg/ml). The slides were examined under a fluorescence microscope and imaged. Approximately 150 cells were digitalized and analyzed from each treatment group using CASP software.
Cell cycle analysis
Cells were plated at a density of 1 x 106 cells/flask in a T25 cm2 flask. After 24 h, PL at the IC25, IC50 and IC75 concentrations was introduced into the respective flasks, and the flasks were incubated for 12 and 24 h. The cells were harvested by trypsinization, and fixed in 80% cold ethanol. The fixed cells were centrifuged and the cell pellets were resuspended in propidium iodide containing RNase. The cell cycle distribution was assessed by a FACSCanto II flow cytometer (Becton-Dickinson, San Jose, CA). PI signals were monitored using a 585/42 band pass filter. The obtained data were analyzed using FACSDiva software.
Immunofluorescence assay (LC-3B detection)
Cells were prepared and labeled according to the manufacturer’s instructions. After 24 h of treatment with PL, the cell culture medium was removed, and cells were washed with PBS and then fixed with paraformaldehyde solution for 15 min. After permeabilization, cells were blocked with FBS-containing buffer for 15 min and then incubated with LC-3B primary antibody solution (rabbit) for 1 h at RT. After washing with Dulbecco’s PBS with 1% Tween-20 wash buffer, staining solution containing the Hoechst dye (1 mg/ml in aqueous) and the DyLight 488 goat anti-rabbit secondary antibody in blocking buffer with FBS was added, and cells were incubated for 1 h at RT protected from light. After washing out the staining solution, the plate was observed and photographed using a fluorescence microscope with the appropriate filter set(s). The approximate absorption/emission maxima of the fluorescent dyes were DyLight 488 (green) conjugates = 493/518 nm and Hoechst dye = 350/461 nm.
Protein interaction network analysis
The quinones and PL-altered gene and protein data were composed from databases and previous literature. To create the gene ontology (GO) enrichment, protein interaction network map and pathway analysis of quinones and PL, we used Stitch (version 4.0) and Cytoscape 3.2 software.
Results
Cytotoxicity studies
An MTT assay was used to measure the cytotoxicity of PL on SiHa cells. The inhibitory concentrations were obtained for 12 and 24 h. The obtained results indicate that PL IC25, IC50 and IC75 values are 5.4 ±1.2 µM, 11.5 ±2.8 µM, and 14.2 ±2.4 µM, respectively. The results revealed that PL treatment decreased the cell viability of SiHa cells.
Morphological analysis
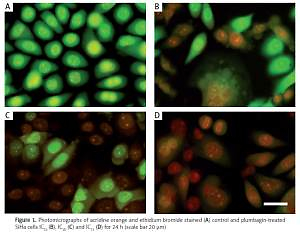
We observed that PL-treated cells were stained yellow to green and unusually fragmented nuclei/chromatin had elegantly expanded and notable features. This result is a definite indication that PL kills the cells through a non-apoptotic process (Figure 1). Here, PL altered the cytosolic morphological features of the SiHa cells, which exhibited the degradation of cytoplasmic components that occurs in autophagocytic vacuoles. The features are consistent with autophagic cell death. Vacuoles were found in PL-treated cells, and the size of the vacuoles enlarged gradually with increased exposure time, and they occupied most of the cytoplasmic region of the cell (Figure 2). The percentage of vacuolated cells increased with an increase in the concentration of PL. The PL treatment resulted in autophagic cells, representing 83%, 71% and 54% of the total cell death population of IC75, IC50 and IC25 treated cells, respectively. Our results clearly indicate that PL triggers cell death through the autophagy pathway in SiHa cells.
Figure 1
Photomicrographs of acridine orange and ethidium bromide stained (A) control and plumbagin-treated SiHa cells IC25 (B), IC50 (C) and IC75 (D) for 24 h (scale bar 20 μm)
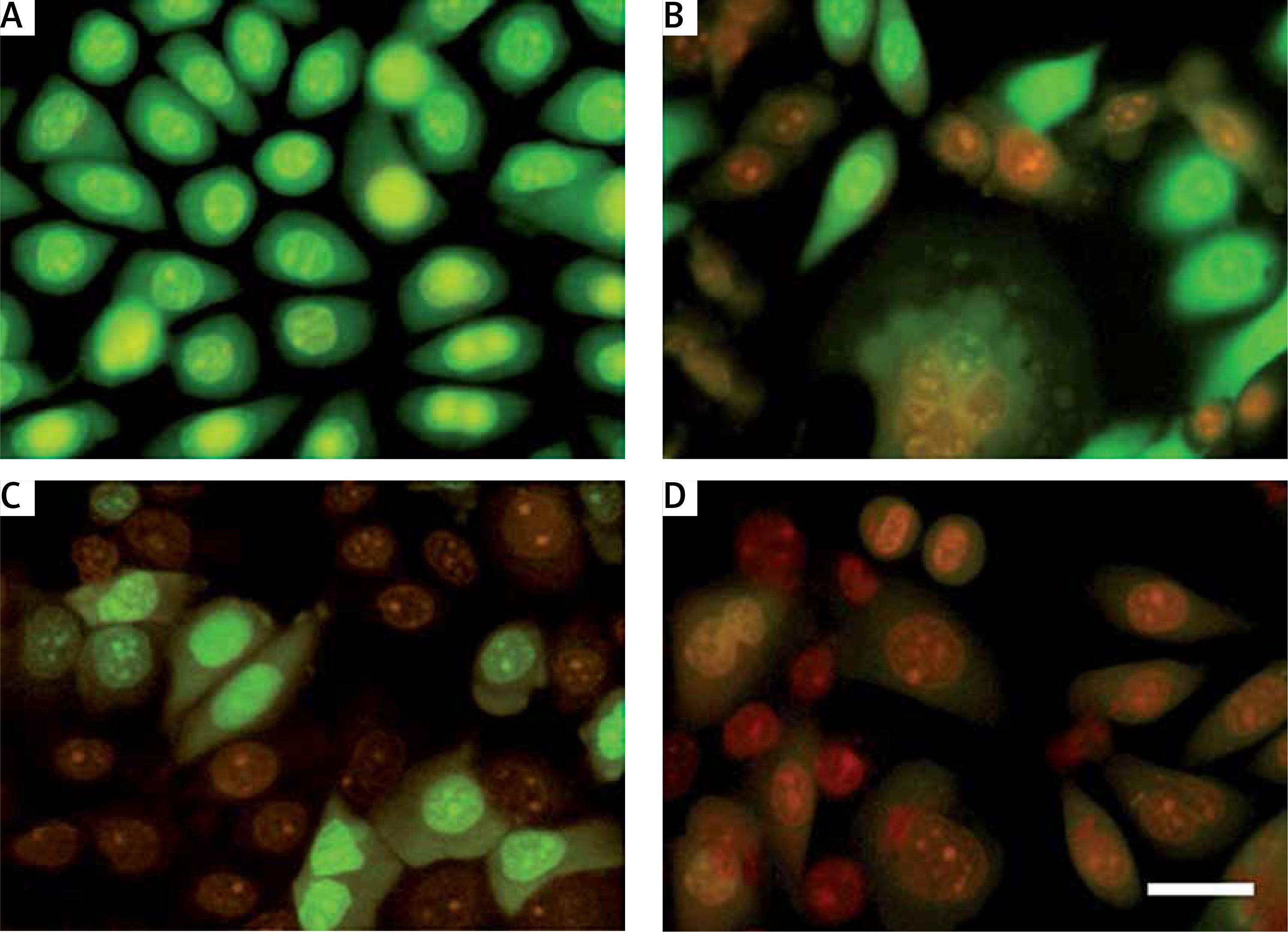
Figure 2
Autophagic morphological and cytological features of IC50 value of plumbagin-treated SiHa cells (A, B). Green and unusual fragmented nuclei/chromatin; C, D – typical marginalization of chromatin, such as bodies, and occurrence of several chromatoid bodies, such as scattered micronucleus; E, F – irreversible cytolysis and membrane disruption with abnormalities in the nuclear structure; G, H – blebbing of the cytoplasm as lobes of pseudopodia. Plumbagin-induced autophagy resulted in ultrastructural changes in the treated cells. As shown, exposure of cells to plumbagin for 24 h caused the presence of autophagocytic vacuoles with degraded cellular material or membranous structures, consistent with the characteristics of cells undergoing autophagy (scale bar 20 μm)
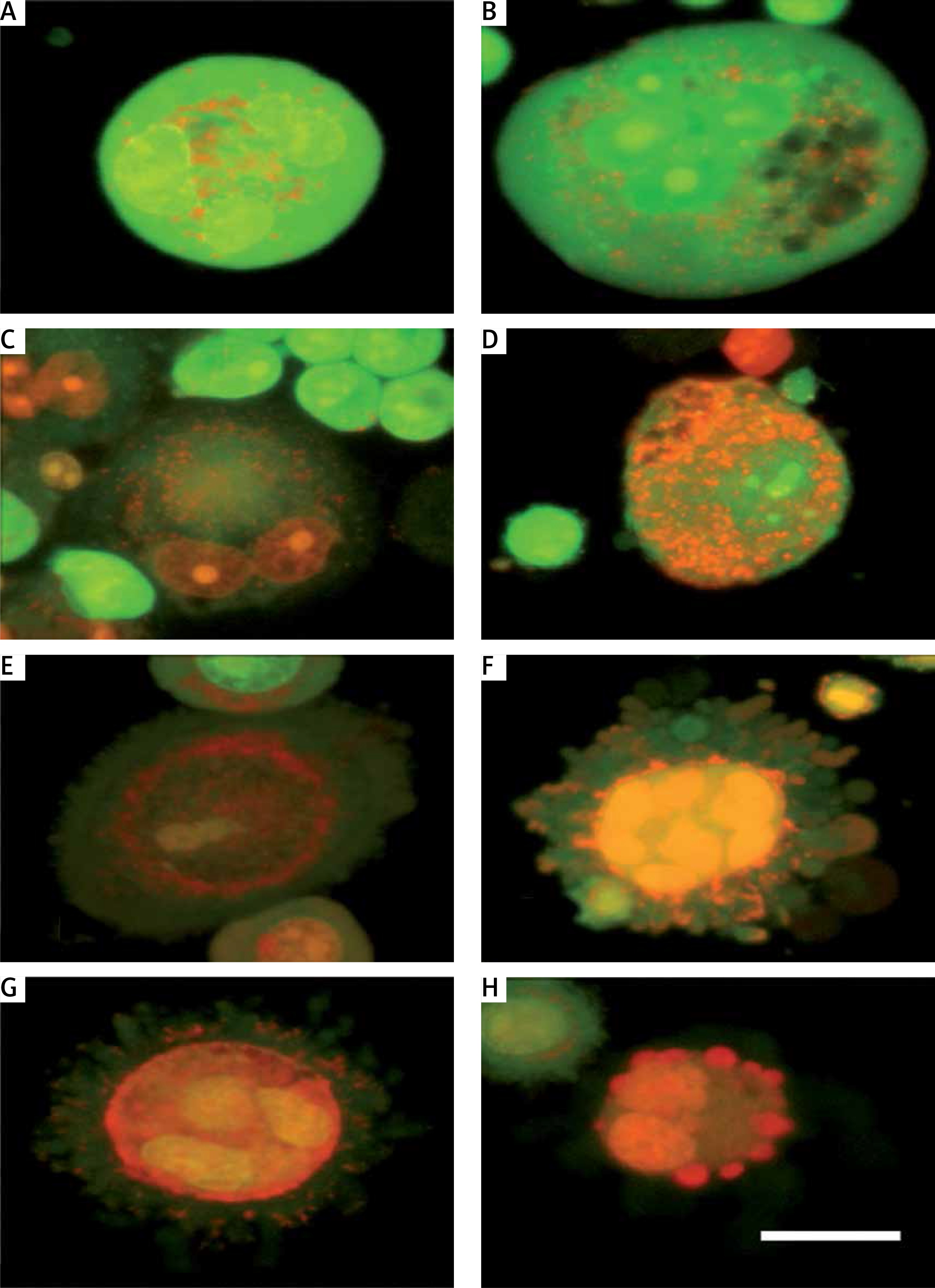
DNA damage analysis
Assessment of DNA strand breaks was analyzed using an alkaline comet assay (Figure 3). Plumbagin treatment triggered DNA fragmentation in SiHa cells, which shows comet-like structure formation by migration of DNA fragments from the nuclei. The PL-untreated control group images indicate intact nuclei that contained undamaged high density DNA with smooth margins. Plumbagin-treatment groups exhibited broom-shaped tails extending from the comet. When compared with the control group, the fluorescence intensity of the comet heads of PL-treated cells was very low. Moreover, the percentage of comet-positive cells and the length of their tails were remarkably increased with increased concentration of PL. Our results suggested that PL enhanced DNA strand breakage in SiHa cells in a concentration-dependent manner.
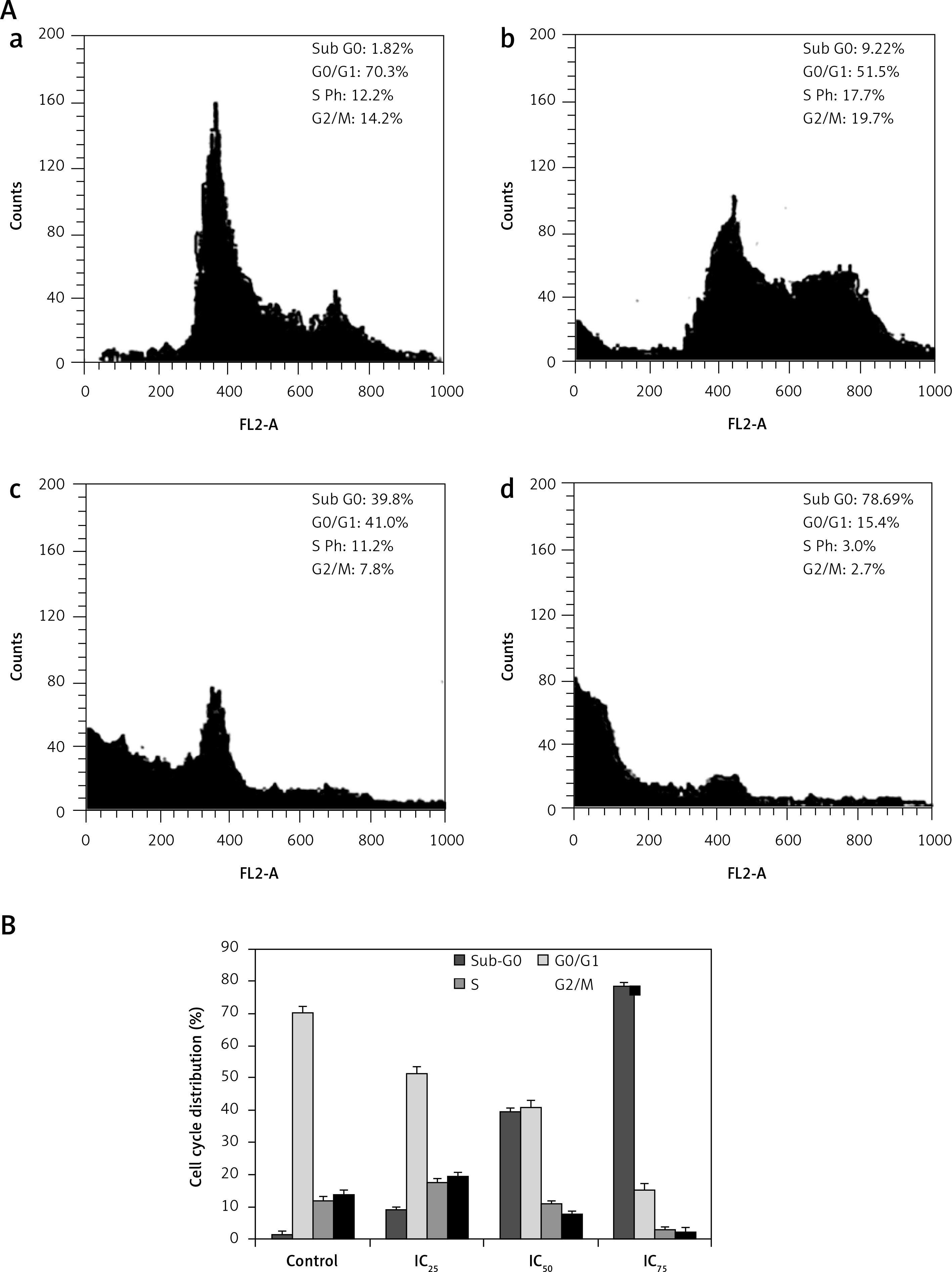
Effect of plumbagin on cell cycle distribution
We observed remarkable changes in the percentage of cells in the sub-G0 (hypodiploid) phase of the cell cycle (Figures 4 and 5). The cell cycle progression results suggested that the proportion of the cell population in sub-G0 phase increased, accompanied by a decrease in the percentage of the cell population in S phase and G2/M phase. When the PL treatment concentration (IC25, IC50 and IC75) and duration (12 and 24 h) increased, the proportion of the cell population in sub-G0 phase also increased. Our results indicate that PL treatment increased the number of hypodiploid cells in a dose- and time-dependent manner. These drastic increases in the percentage of cells in sub-G0 phase clearly revealed that PL induced cell death through DNA damage. The cell cycle progression study results were consistent with the comet assay results.
Figure 4
A – The cell cycle phase was determined by the cellular DNA content measured by flow cytometry of PI stained cells. Cell cycle distribution of plumbagin-treated SiHa cells for 12 h control (a), IC25 (b), IC50 (c) and IC75 (d). Cell cycle distribution is shown as histograms. Percentage data are obtained from two independent experiments. B – Quantitative representation of cell cycle distribution of SiHa cells treated with plumbagin for 12 h
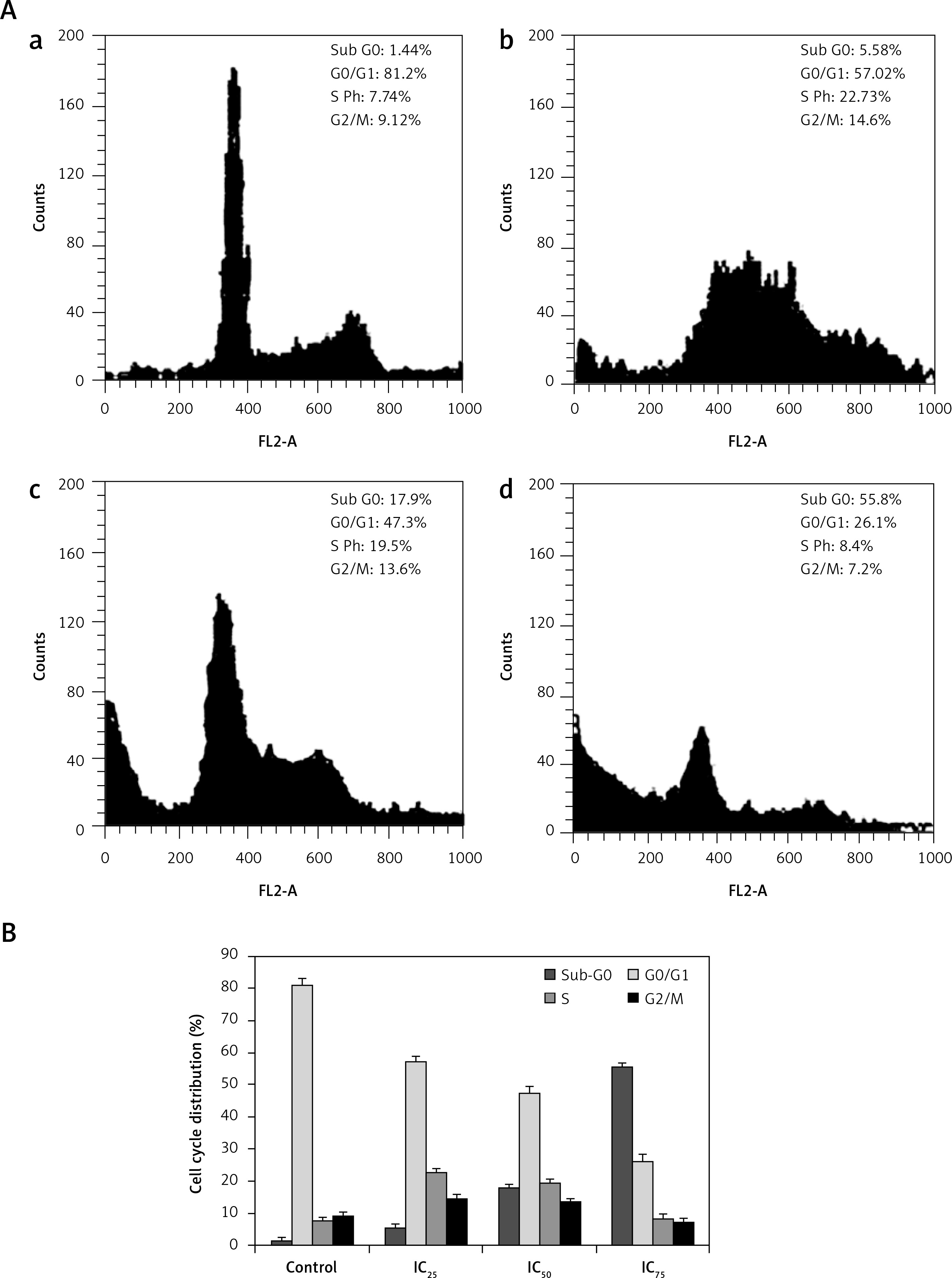
Figure 5
A – Cell cycle distribution of SiHa cells treated with plumbagin for 24 h: control (a), IC25 (b), IC50 (c), IC75 (d). Cell cycle distribution is shown as histograms. Percentage data are obtained from two independent experiments. B – Quantitative representation of cell cycle distribution of SiHa cells treated with plumbagin for 24 h
Immunofluorescence of autophagy marker analysis
By morphological analysis, we found that PL promotes autophagic cell death in SiHa cells. Due to these results we decided to confirm whether PL induces autophagy or not. Thus, the formation and disintegration of autophagosomes were analyzed by immunofluorescence. Microtubule-associated protein 1 light chain 3 (LC3) proteins, such as MAP1-LC3A, MAP1-LC3B and MAP1-LC3C, are widely used autophagy biomarkers. For the duration of autophagy, cytoplasmic LC3-I is split to form the membrane-linked LC3-II, which is involved in autophagosome formation. Figure 6 depicts the immunofluorescence results that indicate an accumulation of autophagosome puncta containing LC3 proteins in PL-treated SiHa cells. In 31% of the PL treated cells, LC-3B protein translocates to the autophagosome from the cytoplasm. As shown in Figure 6, the intensity and number of punctuate LC3B fluorescence are increased after 24 h of treatment with PL. Our results suggested that PL triggered autophagic cell death in SiHa cells.
Figure 6
LC-3B detection in plumbagin-treated SiHa cancer cells as revealed in an immunofluorescence assay. Fluorescence image of cells treated with plumbagin for 24 h (untreated; IC50 dose and IC75 dose: left panel – green color Dylight and right panel – Hoechst dye)
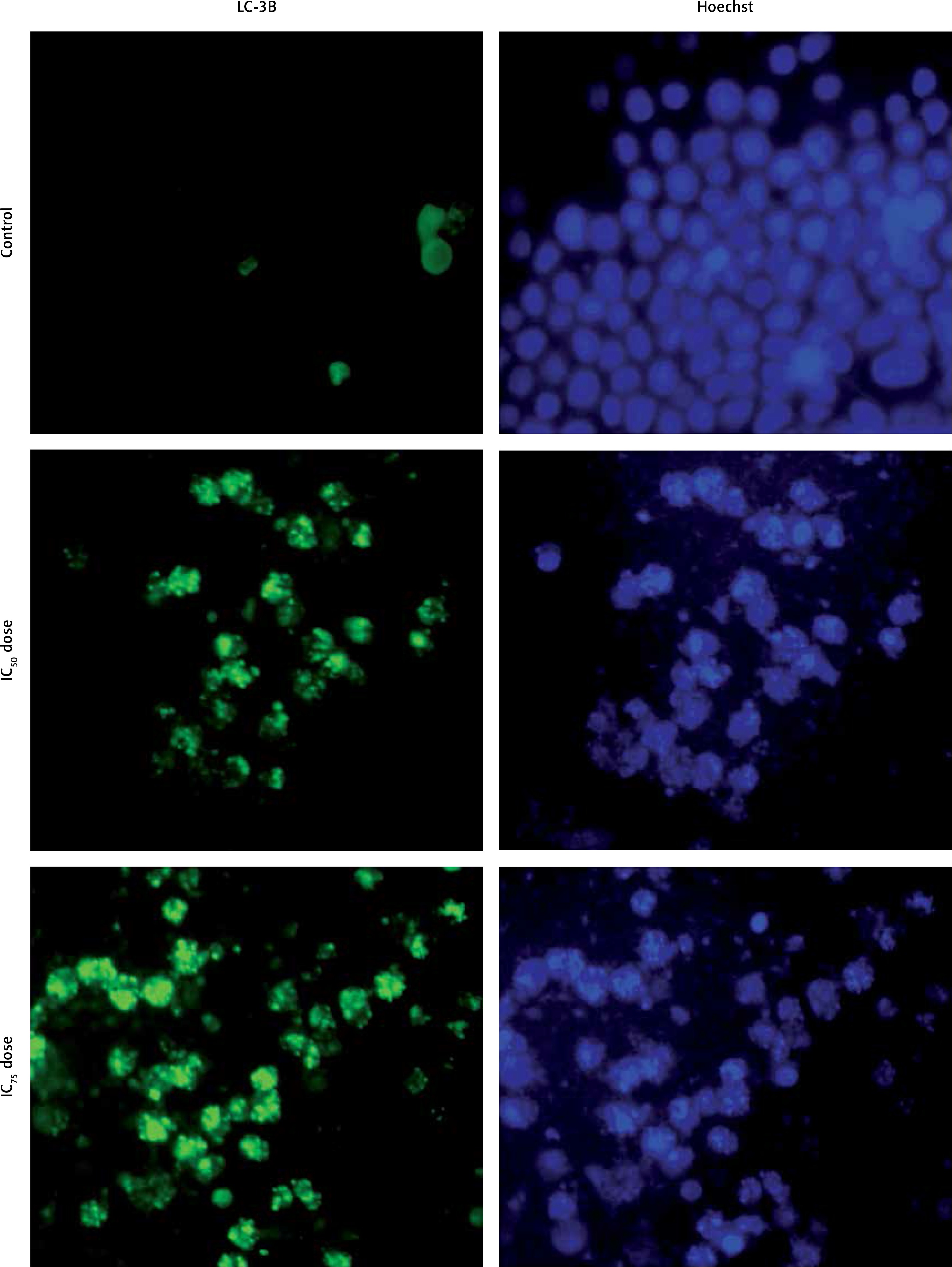
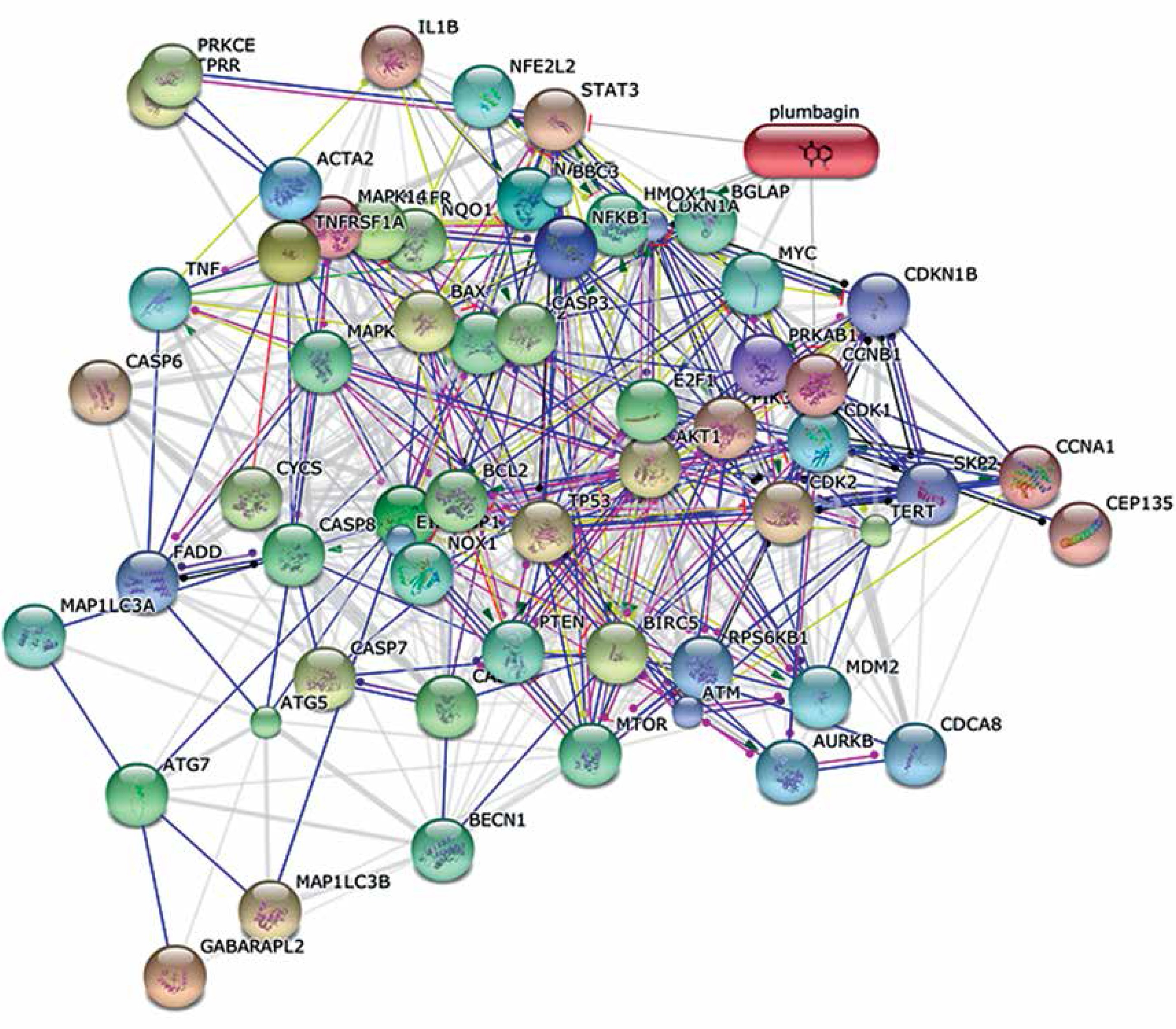
Table I shows protein-protein interaction network analysis of PL. The predicted results clearly indicate that PL has typical physiological interactions with several biological processes, including cellular response to external stimulus, cellular response to abiotic stimulus, cellular response to growth factor stimulus, positive regulation of cell proliferation, aging, neurotrophin TRK receptor signaling pathway, response to abiotic stimulus and regulation of mitotic cell cycle. Also, these pathway specific genes or proteins are involved more often in the cell death mechanism. Figure 7 depicts the predicted proteins and their relationships, including anti-apoptotic genes, pro-apoptotic genes, DNA repair and damage genes, death domain receptors, extracellular apoptotic signals, caspases and regulators and positive and negative apoptotic regulatory genes that are involved. The functional enrichment analysis result of quinine and PL indicates that over 90 genes play a crucial role in modulation of redox signaling dependent/independent pathways.
Table I
Gene ontology (GO) enrichment analysis (biological processes) of plumbagin and data generated from the STITCH (version 4.0) database
Discussion
The redox properties of PL have made it an effective chemotherapeutic agent for human intestinal neoplasia, as well as prostate, lung, cervical, lymphoma, and melanoma cancers, by induction of apoptosis as a target [31–35]. Decades of research has been performed to investigate the apoptosis regulating pathways and to study the development of novel detection and therapeutic modalities in cancer therapy, but recently, the regulation and significance of autophagic cell death, necroptosis and programmed necrosis has become the focus of investigations [4–10]. Earlier studies suggested that PL-treated cells primarily succumbed to apoptotic death [36]. In contrast, considerable events and/or percentages of PL-treated cells suffered non-apoptotic death [27]. The direct mechanisms involved in PL-induced non-apoptotic cell death have remained largely elusive. In the present study, we examined the PL-induced cell morphological changes, DNA damage and cell cycle arrest in the SiHa cell line with special focus on surveillance of the non-apoptotic cell death population.
The cell response to these changes ultimately results in its death, which is neither apoptotic nor non-apoptotic, and detection of DNA damage could be another hallmark diagnostic tool for cell death. The apoptotic signal, either from outside the cell or within, triggers a cascade of molecular events ultimately resulting in cleavage of DNA into fragments. The strand breaks in DNA were detected by comet assay of PL-treated cells, in which the DNA was subjected to the appropriate single cell electrophoretic separation. When the DNA fragments have separated out depending upon their length, the tails will appear as a comet. For a comet to be detected, at least 10–15% of the cells need to have been brought into the scope of apoptosis and the remaining percentage could have undergone other forms of cell death, such as non-apoptotic forms (Figure 2). In such cases, the intact DNA remained as a discrete band on top, and most of the comet morphology observed represented autophagy or necrotic patterns (Figure 3).
Analysis of cell death by any agent, including irradiation, is one of the most reliable indices of the potential application of the compound tested for cancer therapy [37]. The redox system controls the stability of numerous proteins that regulate progression through the cell cycle and other pathways. On the other hand, PL may also induce oxidative stress that might have been linked to the activation of distinct non-apoptotic signaling cascades and might trigger other types of cell death, such as programmed necrosis and autophagy [9, 10]. The direct mechanisms of these non-apoptotic cell deaths have remained largely elusive, and they are a desired end point in cancer therapy [12, 16, 27]. During autophagy, LC-3B protein is translocated from the cytoplasm to the autophagosome, where it is targeted to the lysosome for degradation [38]. In our immunofluorescence results, after PL treatment, the autophagy marker LC-3B exhibited strong fluorescent cytosolic spots, representing autophagosomes, in 31% of cells. Generally, LC-3B plays a normal part in cell growth, development, and cellular homeostasis. Increasing evidence suggests that not just apoptosis but also autophagy can contribute to caspase-independent programmed cell death [5, 15]. Plumbagin might have potential applications in target-based cancer therapy because the mechanism of cell death appears to be essentially non-apoptotic, specifically, occurring through autophagy and necroptosis, which are also one of the desired endpoints in cancer therapy. From this perspective, it would be valuable to confirm the mechanisms of cell killing in the present context by adopting appropriate molecular techniques, including expression of autophagy-related pathways with special focus on redox-mediated cell death signaling.
To understand the interconnection and complexity of pathway networks, the protein-PL interaction was studied using a bioinformatics approach [39]. System-based biological methodologies explore the extensive variety of interconnected gene regulatory networks and the exact molecular mechanisms of their approach to drug resistance, energy metabolism and induced chemotherapy. Network mapping and analysis results suggested that over twelve distinguished pathways contributed through MLD exposure in cancer cells and its resistance. Figure 7 shows the predicted proteins and their relationships. These results suggest that over 90 genes play a crucial role in modulation of redox signaling dependent/independent pathways. Indeed, PL can preclude carcinogenesis by altering the redox enzymes’ expression, cell proliferation regulating genes’ inhibition, and inducing NACD through different mechanisms. Also, the redox-mediated activity of PL might have a potent ability to decrease the progress of drug resistance in cancer cells and reduce toxicity in normal cells.
In conclusion, induction of non-apoptotic cell death through therapeutic interventions is one of the advocated modalities for the therapy of cancer, in addition to induction of cell death through processes such as apoptosis. In this study, it was observed that PL treatment caused cleavage of DNA in SiHa cancer cells, and morphological analysis provided very strong evidence supporting the occurrence of autophagic cell death as a result of PL treatment, but the mechanism of PL-induced cell death in an animal model is remains to be further studied.