Introduction
Congenital or idiopathic hypogonadotropic hypogonadism (CHH or IHH) is a developmental disorder of sex maturation resulting from deficient GnRH release or action. CHH meets the criteria for a rare disease with an estimated prevalence of 1–10 cases per 100,000 births [1]. The condition manifests as absent or delayed puberty and infertility with low serum levels of gonadotropins: follicle-stimulating hormone (FSH) and luteinising hormone (LH). In about 50–60% cases, in addition to CHH, there is an impairment of the sense of smell [2]. The combination of anosmia and hypogonadotropic hypogonadism is referred to as Kallmann syndrome (KS). This unique phenotype is a result of shared embryonic developmental signalling pathways of GnRH and olfactory neurons. GnRH neurons originate in the nasal placode at an early stage of embryo development and then migrate along vomeronasal nerves towards the cribriform plate and into the forebrain to finally reach the hypothalamus by the time of birth [3]. Alterations in this migratory process or loss of GnRH neurons impair sexual development and reproductive competence. GnRH is a key factor in human reproduction, impacting both natural and assisted fertility [4, 5].
CHH is not only clinically heterogeneous, but is also even more complex in terms of genetic variability and inheritance patterns. High-throughput technologies have more recently allowed the rate of genetic discovery to greatly accelerate. To date, almost 40 genes have been found to play a role in Kallmann syndrome and normosmic congenital hypogonadotropic hypogonadism (nCHH) pathogenesis, but only up to 40% of cases can be explained by their mutations [6].
The list of CHH-associated genes is still expanding, with the latest report on AMH and AMHR2 in CHH and NDNF mutations exhibiting deleterious effects in Kallmann syndrome [7, 8]. While most CHH mutations are inherited as X-linked recessive, autosomal dominant, or autosomal recessive modes, some genes are shown to exert synergistic heterozygosity. Oligogenic, mostly digenic, interaction was identified for several CHH gene partners, among others: KAL1/PROKR2 [9], FGFR1/GNRHR [10], FGFR1/NELF [10, 11], FGFR1/FGF8 [12], KAL1/WDR11 [13], and SEMA3A/FGFR1 [14]. The overall prevalence of digenic mutations in CHH patients varies in different studies, but it is reported at about 10% [13–15]. Interestingly, in more than one-third of cases a single gene mutation is identified, thus it can be expected that the true frequency of oligogenicity is much higher [15].
Given that the complex architecture underlying idiopathic hypogonadotropic hypogonadism is still largely unknown, our study used targeted next-generation sequencing (NGS) to screen for DNA variants in a cohort of CHH/KS patients. Data on monogenic variants in ANOS1 and FGFR1, the classical CHH genes, were recently published by our group [16]. Here we report the identification of new oligogenic combinations of variants. All recognised genes are known to contribute to the phenotype of the Kallmann syndrome and normosmic congenital hypogonadotropic hypogonadism, but our results point to potential new “partners” underlying digenic and trigenic patterns.
Material and methods
Patients
Forty-seven patients (31 males and 16 females, 25 nCHH and 22 KS) were enrolled for the study. They were referred to the Department of Genetics for participation in genetic studies based on diagnosis of CHH. Inclusion clinical criteria were as follows: 1) failure to enter spontaneous puberty by the age of 16 years in girls and by the age of 18 years in boys, or pharmacologically induced puberty below this age; 2) cryptorchidism and/or micropenis in male neonates; and 3) infertility and decreased libido. Absent or delayed puberty was the most common indication (28 patients), cryptorchidism and/or micropenis diagnosed at minipuberty was reported in 16 patients, and diagnosis of CHH due to infertility investigation was reached in 3 patients. Enrolled individuals showed low testosterone or oestradiol levels and low or normal LH and FSH concentrations. Patients with other associated pituitary deficiencies or abnormal findings on diagnostic imaging of the hypothalamic-pituitary region were excluded from the present study. Olfactory function was assessed by formal testing or where unavailable by self-reporting by the patients. All patients (or in the case of participants under 16 years of age, their parents/guardians) signed an informed consent form for publication of the study results. All research and methods were performed in accordance with relevant regulations as approved by the Bioethics Committee of the Polish Mother’s Memorial Hospital Research Institute (approval number 42/2012). All the procedures followed the principles of the Declaration of Helsinki.
Targeted DNA sequencing and bioinformatic analyses
DNA was automatically extracted from peripheral blood leukocytes using the MagCore Genomic DNA Whole Blood Kit (RBC Bioscience, Taiwan, Cat. No. MGB400-02-SP), according to the manufacturer’s instructions. Targeted NGS, alignment, and variant calling were performed as previously described [16]. The customised panel included 51 CHH-associated and candidate genes: ANOS1, AXL, CCDC141, CGA, CHD7, DUSP6, EBF2, FEZF1, FGF17, FGF8, FGFR1, FLRT3, FSHB, FSHR, GNRH1, GNRHR, HESX1, HS6ST1, IL17RD, KISS1, KISS1R, LEP, LEPR, LHB, LHCGR, LHX3, LHX4, NSMF, NR5A1, NROB1, PCSK1, PNPLA6, POLR3B, PROK2, PROKR2, PROP1, SEMA3A, SEMA3E, SEMA7A, SOX10, SOX2, SPRY4, SRA1, STUB1, TAC3, TACR3, TSHZ1,TUBB3, VAX1, VCX3A, and WDR11.
Probes for the targeted regions were designed using the Illumina Design Studio on-line software (https//designstudio.com) and finally contained 1070 amplicons, 175 bp length, with 99% sequencing coverage. The DNA of all samples was quantified using a Quantus Fluorometer (Promega, WI, USA) and diluted to desired 10–25 ng/μl concentration. All steps of the library preparation, including oligos hybridisation, ligation, amplification, clean-up, and normalisation libraries, were performed using the DNA Amplicon Assay v2 kit (Illumina, Cat. No. FC-134-2002) according to the TrueSeq Custom Amplicon Low Input Library Prep Reference Guide (Illumina, CA, USA) and relevant protocols.
PCR products barcoded with TruSeq Custom Amplicon Index Kit (Illumina, Cat. No. 2 FC-130-1003) were pooled, loaded into the cartridge (MiniSeq High Output Kit, 300 cycles, Illumina, Cat. No. FC-420-1004), and submitted to the Illumina MiniSeq platform (Illumina, CA, USA) using 2 × 150 bp paired-end runs.
Data obtained from the instrument were pre-processed in the built-in software. Reads were aligned to the human reference genome (version GRCh37). Variants were filtered according to alternative read depth, population frequency (ExAC, 1000 Genomes, GnomAD), prediction programs (Mutation Taster, SIFT, PolyPhen, NNSplice, DANN, GERP, PROVEAN), and available databases (dbSNP, HGMD, ClinVar). Variants were classified according to American College of Medical Genetics 2015 recommendations.
Validation by Sanger sequencing
Sanger sequencing was used to verify selected class 3, 4, and 5 variants. Primers were designed to anneal to upstream and downstream of DNA regions containing selected variants using Primer3 Input (version 0.4.0) program. After PCR amplification with PCR Master Mix (Promega, Cat. No. M7423), products were cleaned and sequenced using BigDye Terminator v3.1 Cycle Sequencing Kit (ThermoFisher Scientific, Cat. No. 4337455) on a 3500 Genetic Analyzer instrument (Thermo Fisher Scientific, MA, USA). All novel variants and those reported in CHH patients for the first time were submitted to ClinVar [17].
Control group
Variants were checked in 100 healthy adult controls and 120 in-house exomes. Additionally, to maximise the volume of general population data, we utilised four widely used control databases: (1) gnomAD (n = 141,456, http://gnomad.broadinstitute.org/), (2) ExAC (n = 60,706, http://exac.broadinstitute.org/), (3) NHLBI Exome Sequencing Project (n = 6,503, http://evs.gs.washington.edu/EVS/), and (4) 1000 Genomes Project Phase 3 database (1000 G; n = 2,504, http://www.internationalgenome.org/).
Results
Patient characteristics
Forty-seven patients, including 25 with congenital hypogonadotropic hypogonadism without anosmia and 22 with Kallmann syndrome, underwent customised CHH gene panel screening. In 16 patients, primary clinical symptoms were cryptorchidism and/or micropenis, in 28 cases absent/delayed puberty, and in 3 cases infertility and/or decreased libido. None of the probands had a positive family history of CHH. All patients were Caucasians of Polish origin (Table I).
Table I
Variants identified in patients with normosmic CHH and Kallmann syndrome
| Patient no. (sex) | 1 (M) | 2 (M) | 3 (F) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clinical diagnosis (symptoms) | nCHH (absent puberty) | KS (cryptorchidism and micropenis at birth, anosmia) | KS (delayed puberty, primary amenorrhea, anosmia) | ||||||||||||
| Patient’s genotype | SPRY4:c.[722C>A];[=] | SEMA3A: c.[1303G>A][=] | SEMA7A:c.[618_619delCC];[=] | CHD7: c.[2840G>A][=] | SEMA7A:c.[916G>A];[=] | SRA1: c.[377G>A][=] | |||||||||
| Gene | SPRY4 | SEMA3A | SEMA7A | CHD7 | SEMA7A | SRA1 | |||||||||
| Exon/intron | Exon 3 | Exon 11 | Exon 6 | Exon 11 | Exon 8 | Exon 3 | |||||||||
| Reference sequence | NM_030964.3 | NM_006080.2 | NM_003612.3 | NM_017780.3 | NM_003612.4 | NM_001035235.3 | |||||||||
| Nucleotide variant | c. 722C>A | c. 1303G>A | c.618_619delCC | c.2840G>A | c.916G>A | c.377G>A | |||||||||
| Protein variant | p.Ser241Tyr | p.Val435Ile | Ile206Metfs*5 | p.Arg947Gln | p.Val306Ile | p.Arg126His | |||||||||
| Variant orygin | Maternal | Paternal | Maternal | Paternal | Maternal | Excluded in maternal sample, paternal DNA n/a | |||||||||
| Pathogenicity based on in silico prediction tools | Pathogenic: PolyPhen, SIFT, DANN, GERP, LRT, MutationTaster, PROVEAN Benign:dbNSFP.FATHMM, MetaLR, MetaSVM and MutationAssessor | Pathogenic: SIFT, DANN, GERP, LRT, MutationAssessor MutationTaster Benign: PolyPhen, dbNSFP.FATHMM, MetaLR, MetaSVM, PROVEAN | Pathogenic: Mutation Taster benign: – | Pathogenic: PolyPhen, MutationTaster DANN, GERP, dbNSFP.FATHMM, LRT, MetaLR, MetaSVM, MutationAssessor, PROVEAN Benign: – | Pathogenic: : PolyPhen, DANN, GERP, MutationTaster Benign: SIFT,dbNSFP.FATHMM, LRT, MetaLR, MetaSVM, MutationAssessor and PROVEAN | Pathogenic: : SIFT, DANN, GERP, MutationAssessor and MutationTaster Benign: PolyPhen, dbNSFP.FATHMM, LRT, MetaLR, MetaSVM and PROVEAN. | |||||||||
| MAF: | |||||||||||||||
| ExAC | 0.00458 | 0.014 | 0.00001 | 0.00011 | 0.00028 | 0 | |||||||||
| 1000Genomes | 0,002 | 0,002 | 0.00001 | 0,00007 | 0 | ||||||||||
| gnomAD | 0.00449 | 0.01367 | 0.000004061 | 0.00005 | 0.00026 | 0 | |||||||||
| TOPMED | 0.00401 | 0.01215 | 0.00001 | 0.00006 | 0.00003 | ||||||||||
| Variant in databases | HGMD: CM133833 (Kallmann syndrome) | HGMD: CM144399 (Hirschsprung disease) | HGMD: – | HGMD: – | HGMD: – | HGMD: – | |||||||||
| dbSNP: rs139512218 | dbSNP: rs147436181 | dbSNP: rs761366929 | dbSNP: rs768481542 | dbSNP: rs563651407 | dbSNP: rs1306465994 | ||||||||||
| ClinVar: RCV000454560.1 (Uncertain significance* – not specified), RCV000043616.4 (Risk factor – Hypogonadotropic hypogonadism 17 with or without anosmia). | ClinVar: RCV000032921.2 (Risk factor – Hypogonadotropic hypogonadism 16 with or without anosmia), RCV000455242.1 (Benign* – not specified), RCV000059788.1 (Not provided – not provided). | Submitted, ClinVar Submission number SCV000930001 | ClinVar: RCV000634441.1 (Uncertain significance* – CHARGE association) | Submitted, ClinVar Submission number SCV000930002 | Submitted, ClinVar Submission number SCV000930003 | ||||||||||
| Variant type and class** | Missense substitution VUS, class 3 | Missense substitution Likely pathogenic, class 4 | Two nucleotides deletion Likely pathogenic, class 4 | Missense substitution VUS, class 3 | Missense substitution VUS, class 3 | Missense substitution VUS, class 3 | |||||||||
| Patient no. (sex) | 4 (F) | 5 (M) | |||||||||||||
| Clinical diagnosis (symptoms) | nCHH (absent puberty, primary amenorrhea) | KS (delayed puberty, anosmia) | |||||||||||||
| Patient’s genotype | CCDC141:c.[2299G>A];[=] | POLR3B:c.[1244T>C];[2570+1G>A] | PROKR2:c.[889G>T];[=] | SPRY4:c.[530A>G];[=] | NSMF: c.[1261C>T];[=] | ||||||||||
| Gene | CCDC141 | POLR3B | PROKR2 | SPRY4 | NSMF (NELF) | ||||||||||
| Exon/intron | Exon 15 | Exon13 | Intron22 | Exon 2 | Exon3 | Exon13 | |||||||||
| Reference sequence | NM_173648.3 | NM_018082.5 | NM_018082.5 | NM_144773.2 | NM_030964.3 | NM_001130969.1 | |||||||||
| Nucleotide variant | c.2299G>A | c.1244T>C | c.2570+1G>A | c.889G>T | c.530A>G | c.1261C>T | |||||||||
| Protein variant | p.Asp767Asn | p.Met415Thr | p.Val297Phe | p.177LysArg | p.Leu421Phe | ||||||||||
| Variant orygin | Maternal | Paternal | Maternal | Paternal | Maternal | Maternal | |||||||||
| Pathogeneicity based on in silico prediction tools | Pathogenic SIFT, PolyPhen, DANN, GERP, MutationAssessor, MutationTaster Benign:dbNSFP.FATHMM, LRT, MetaLR, MetaSVM, PROVEAN | Pathogenic: SIFT,DANN, GERP, LRT, MutationAssessor, MutationTaster, PROVEAN Benign: PolyPhen, dbNSFP.FATHMM, MetaLR, MetaSVM | Pathogenic: DANN, GERP, MutationTaster, scSNV-splicing Benign: – | Pathogenic:SIFT, PolyPhen, DANN, GERP, LRT, MutationAssessor, MutationTaster, PROVEAN Benign: dbNSFP.FATHMM, MetaLR | Pathogenic: PolyPhen, DANN, GERP, LRT, MutationAssessor, MutationTaster Benign: SIFT,dbNSFP.FATHMM, MetaLR, MetaSVM, PROVEAN | Pathogenic: SIFT, PolyPhen, DANN, GERP, LRT, MutationTaster, PROVEAN Benign: dbNSFP.FATHMM, MetaLR, MetaSVM, MutationAssessor | |||||||||
| MAF: | |||||||||||||||
| ExAC | 0.00392 | 0.00062 | 0.00002 | 0 | 0.00160 | 0 | |||||||||
| 1000Genomes | 0.00040 | 0.00020 | – | 0 | 0.001 | 0 | |||||||||
| gnomAD | 0.00042 | 0.00056 | 0.00005 | 0 | 0.00162 | 0 | |||||||||
| TOPMED | 0.00033 | 0.00065 | 0.00004 | – | 0.00135 | – | |||||||||
| Variant in databases | HGMD: – | HGMD: – | HGMD: CS131199 | HGMD: – | HGMD: CM133831 (KS) | HGMD: – | |||||||||
| dbSNP:rs141939661 | dbSNP:rs199504211 | dbSNP:rs753943393 | dbSNP: – | dbSNP: rs78310959 | dbSNP: – | ||||||||||
| Submitted, ClinVar Submission number SCV000930004 | ClinVar: RCV000372557.2 (Uncertain significance** – not specified) | ClinVar: RCV00760972 (Hypomyelinating Leucodystrophy 8, with or without HH | Submitted, ClinVar Submission number SCV000930005 | ClinVar: RCV000356060.1 (Uncertain significance* – not specified), RCV000043614.3 (Pathogenic – HH 17 with or without anosmia). | Submitted, ClinVar Submission number SCV000930006 | ||||||||||
| Variant type and class* | Missense substitution VUS, class 3 | Missense substitution VUS, class 3 | Splice donor Pathogenic , class 5 | Missense substitution VUS, class 3 | Missense substitution VUS, class 3 | Missense substitution VUS, class 3 | |||||||||
Oligogenic findings based on NGS custom panel screening
Patients were screened with the custom NGS panel with a mean read depth of 1142x and 90.3% coverage of the target region. Herein we report of patients with at least two heterozygous variants identified. A total of 12 variants were confirmed by Sanger sequencing in five individuals (Figure 1). Variants were found in SPRY4 (2 patients), SEMA7A (2 patients), POLR3B (2 variants in 1 patient), and SEMA3A, CHD7, PROKR2, CCDC141, NSMF, and SRA1 (each in a single patient). Gene coordinates, chromosomal localisation, and interactions are shown in Figure 2. Clinical phenotypes, characteristics of patients, and reported variants are detailed below.
Figure 1
Targeted sequencing identifies oligogenic variants in CHH patients. A – Segregation of identified variants within families. Probands/affected individuals are identified by the arrow. Circles denote females; squares denote males. M: mutation; NT: not tested. B – Sequence chromatograms of affected patients. Family 1: SPRY4: c.722C>A and SEMA3A: c. 1303G>A; M2 chromatogram identifies both a rare variant and a common polymorphism rs7804122 (the dotted line arrow). Family 2: SEMA7A: c. 618_619delCC and CHD7: c. 2840G>A. Family 3: SEMA7A: c. 916G>A and SRA1: c. 377G>A. Family 4: CCDC141: c. 2299G>A and POLR3B: c. 1244T>C; 2570+1G>A. Family 5: PROKR2:c. 889G>T, SPRY4:c. 530A>G, and NSMF: c. 1261C>T. The site of mutation is identified by the arrow
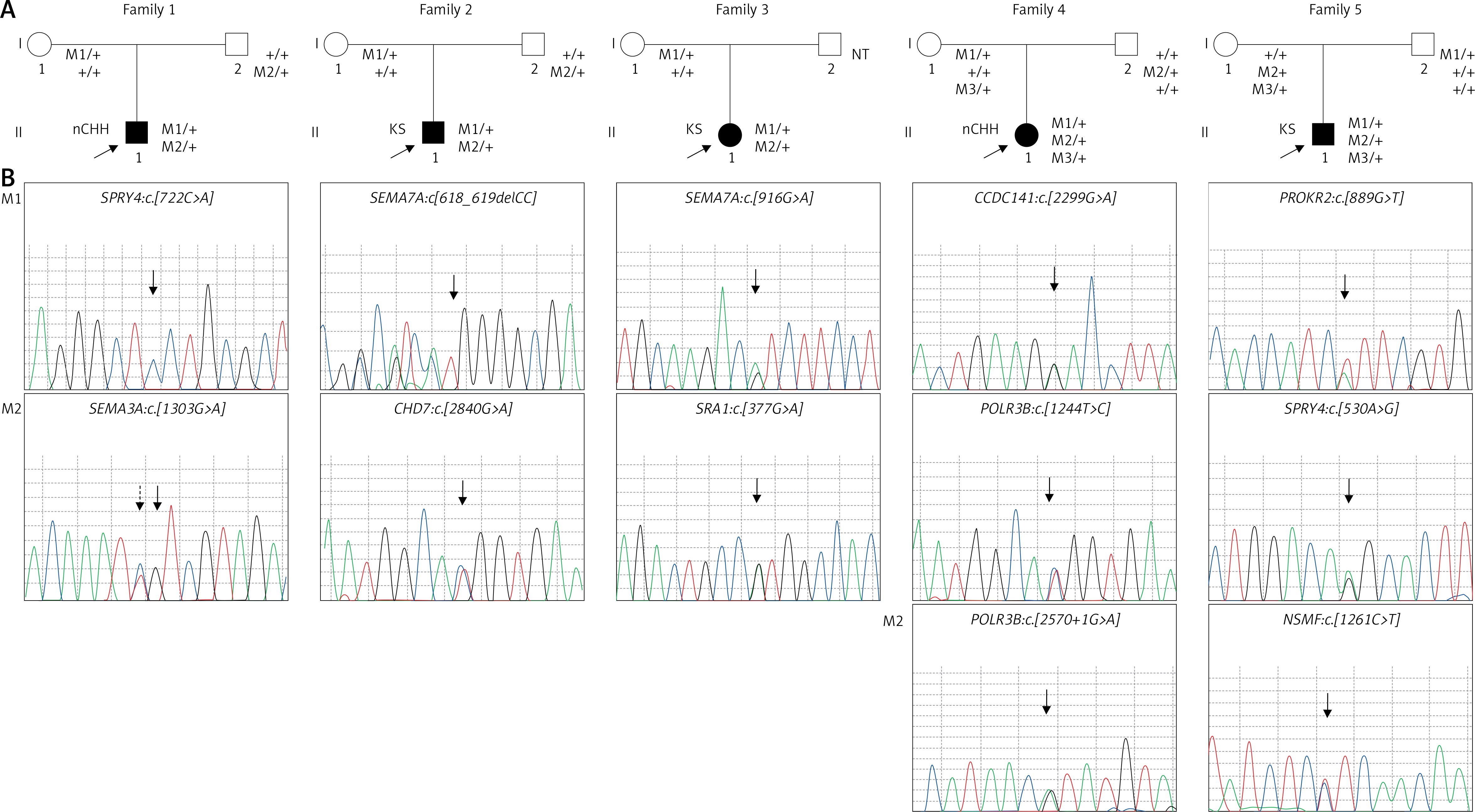
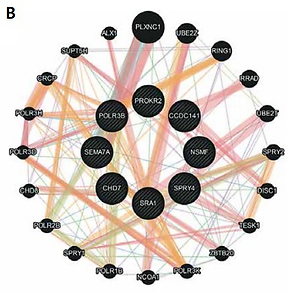
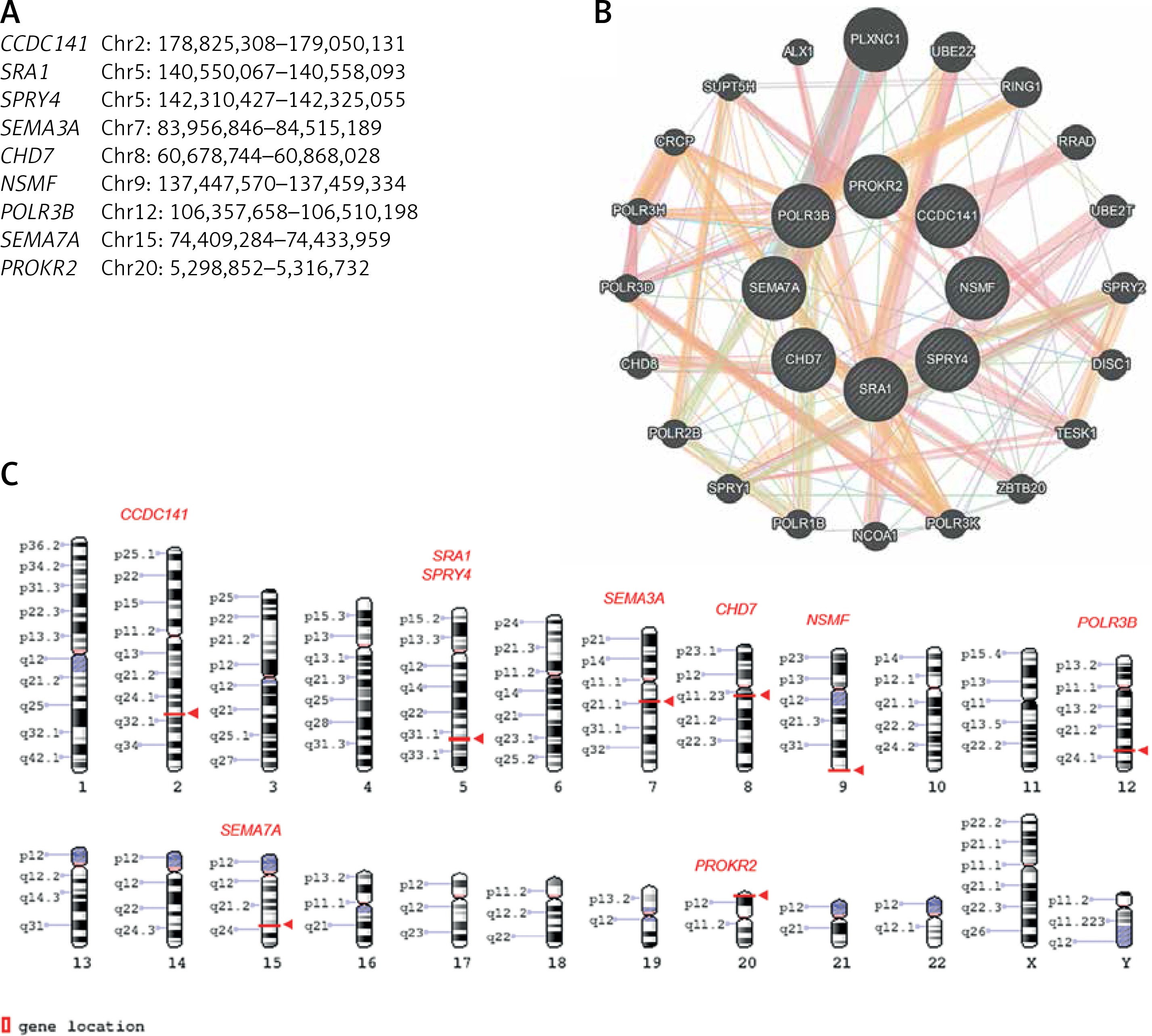
Figure 2
Genes linked to oligogenic inheritance in the studied CHH cohort. A – List of genes with genomic coordinates for the GRCh38 reference assembly. B – Gene-gene interaction based on online tool https://genemania.org/. C – Chromosomal localisation of genes of interest based on https://www.ncbi.nlm.nih.gov/genome/tools/gdp
Identified variants
SPRY4
Two rare missense SPRY4 (MIM 607984) variants were identified in the studied cohort of CHH patients. SPRY4 c.722C>A (p.Ser241Tyr) was found in a man aged 28 years (patient 1) with a clinical diagnosis of normosmic CHH. The patient underwent clinical evaluation at the age of 24 years because of decreased libido and infertility. Variant SPRY4 c.722C>A (p.Ser241Tyr) is predicted to be deleterious by PolyPhen, SIFT, DANN, GERP, LRT, MutationTaster, and PROVEAN. Another rare variant SPRY4 c.530A>G (p.Lys177Arg) was identified in a 19-year-old male (patient 5) born with underdeveloped male genitalia diagnosed with Kallmann syndrome at the age 13 years. Laboratory studies revealed low gonadotropins with minimal increase after GnRH stimulation. The patient was treated with gonadotropins with no effect. Subsequently testosterone therapy was initiated, and development of secondary sexual characteristics was partially achieved. The identified mutation is predicted as pathogenic by PolyPhen, DANN, GERP, LRT, MutationAssessor, and MutationTaster, while as benign by SIFT and PROVEAN. Both mutations are categorised as variants of uncertain significance (VUS) but were previously reported in patients with oligogenic CHH [15, 18].
SEMA3A
Missense variant SEMA3A (MIM 603961) c.1303G>A (p.Val435Ile) was identified in patient 1 with normosmic CHH. The variant is predicted to be pathogenic by SIFT, DANN, GERP, LRT, MutationAssessor, and MutationTaster and benign by PolyPhen and PROVEAN. It reached class 4 in variant classification (likely pathogenic) and was reported in Kallmann syndrome [14].
CHD7
Missense mutation CHD7 (MIM 608892) c.2840G>A, which predicts substitution p.(Arg947Gln), was identified in a 20-year-old male (patient 2), who was at birth diagnosed with cryptorchidism and micropenis. He underwent orchidopexy at the age of 2 years. At the age of 11 years a diagnosis of Kallmann syndrome was established based on anosmia, other clinical symptoms, and laboratory test results. Because there was no response on gonadotropin therapy, testosterone injections were initiated at the age of 13 years. Development of secondary sexual characteristics was partially achieved. The patient reported decreased libido; a semen analysis was not performed. The family history is remarkable because the father’s brother was born with choanal atresia and died in the first days of life. The variant is classified as pathogenic by all used prediction programs: PolyPhen, MutationTaster DANN, GERP, dbNSFP.FATHMM, LRT, MetaLR, MetaSVM, MutationAssessor, and PROVEAN. According to accepted criteria it was marked as class 3, a variant of uncertain significance (VUS). Interestingly, the variant was previously reported in a single case of CHARGE syndrome [19].
SEMA7A
Analysis of the sequencing data indicated two SEMA7A (MIM 607961) variants in the tested group of patients. A novel frameshift mutation of SEMA7A c.618_619delCC was identified in KS proband (patient 2). The variant was not found in 100 healthy adult controls and 120 in-house exomes. The minor allele frequency was 0.00001 in both ExAC and 1000 genomes. MutationTaster predicts the variant to be disease causing. This deletion is expected to cause a frameshift and a subsequent premature stop codon. The novel variant was submitted to ClinVar and was assigned with accession number, SCV000930001. Another SEMA7A variant was found in a 22-year-old female (patient 3) diagnosed with KS based on delayed puberty, primary amenorrhoea, and anosmia. At the age of 14 years, replacement hormone therapy was initiated and secondary sexual characteristics were achieved. Missense SEMA7A c.916G>A (p.Val306Ile) variant is predicted to be disease causing by MutationTaster, tolerated by SIFT, and possibly damaging by PolyPhen. Following ACMG–AMP recommendations, it was classified as class 3, variant of uncertain significance (VUS). The variant occurs within a conserved sequence and was not found in 100 healthy adult controls and 120 in-house exomes. The minor allele frequency in ExAC database is 0.0003. The variant was never reported in CHH. Following submission to ClinVar it was assigned with the accession number, SCV000930002.
SRA1
Sequencing data revealed SRA1 (MIM 603819) variant c.377G>A (p.Arg126His) in patient 3 diagnosed with Kallmann syndrome. SRA1 c.377G>A variant potentially affects both core SRA RNA and protein SRAP. At the protein level it is predicted to be disease causing by Mutation Taster, but classified as tolerated and benign by SIFT and PolyPhen, respectively. The mutation occurs within conserved sequence, and it was not found in 100 healthy adult controls and 120 in-house exomes. The minor allele frequency was 0 for SRA1 in the ExAC database. Based on the used criteria it was classified as variant of uncertain significance (VUS). The novel variant was submitted to ClinVar and was assigned with the accession number SCV000930003.
CCDC14
In a single case, analysis of the NGS panel sequencing data indicated a missense mutation (c.2299G>A) in CCDC141 gene (MIM 617147) that predicts Asp to Asn substitution at residue 767. The variant was qualified as disease causing or deleterious by Mutation Taster and SIFT, respectively. PolyPhen predicts the variant to be possibly damaging. Based on the used criteria it was assigned to class 3, variant of uncertain significance (VUS). The variant was submitted to ClinVar and was assigned with the accession number, SCV000930004. A 26-year-old female (patient 4) harbouring the variant was diagnosed with HH at the age of 18 years due to absent puberty and primary amenorrhoea. She was reported to have normal neurologic exam. Development of secondary sexual characteristics was induced upon hormone replacement therapy. After discontinuation of the therapy at the age of 23 years, amenorrhoea returned.
POLR3B
Two variants in the POLR3B gene (MIM 614366): T-to-C transition c.1244T>C leading to an amino acid change p.(Met415Thr) and an intronic mutation at a donor site c.2570+1G>A were identified in patient 4 presenting with normosmic CHH. In silico, the c.1244T>C (p.Met415Thr) variant was predicted to be deleterious with Mutation Taster and SIFT, but benign with PolyPhen. The variant was previously reported in patients presenting with CHH [20]. Mutation at the donor site c.2570+1G>A shows no records in ExAC, 1000 genomes; nor was it found in 120 in-house exomes. According to NNSPLICE, the mutation creates a splice defect and was assigned as pathogenic, class 5.
PROKR2
Targeted resequencing with NGS panel revealed a missense mutation c.889G>T (p.Val297Ile) in PROKR2 gene (MIM 607123). This rare variant was identified in a 19-year-old male (patient 5) diagnosed with Kallmann syndrome. The variant was predicted to be deleterious by Mutation Taster, SIFT, PolyPhen, and PROVEAN. It shows no records in ExAC, 1000 genomes and was not found in 120 in-house exomes. Based on the used criteria, it was classified as variant of uncertain significance (VUS). Following submission to ClinVar, the novel variant was assigned with the accession number SCV000930005.
NSMF
We identified a novel variant c.1261C>T in the coding sequence of NSMF gene (formerly NELF) (MIM 608137) in a subject diagnosed with KS (patient 5). The variant predicts leucine to phenylalanine substitution (p.Leu241Phe). MutationTaster, SIFT, and PolyPhen assigned this substitution as deleterious. The variant was not previously reported, shows no records in ExAC, 1000 genomes, and was not found in 120 in-house exomes. According to accepted criteria it was labelled as class 3, variant of uncertain significance (VUS). The novel variant was submitted to ClinVar and was assigned with the accession number SCV000930006.
Oligogenic patterns
In the tested cohort, comprising 47 CHH subjects, 5 patients presented with rare variants with pathogenicity ranging from class 5 (pathogenic) to class 3 (VUS) in more than one gene. All variants were revealed in genes implicated in oligogenic CHH, but contributing genes were never reported in the combinations we describe here. Thus, our results point to potential new gene “partners” possibly playing synergistic roles in digenic/trigenic inheritance.
Digenic variants identified in Patient 1 (SPRY4:c.[722C>A];[=], SEMA3A: c.[1303G>A][=]) and Patient 2 (SEMA7A:c.[618_619delCC];[=], CHD7: c.[2840G>A][=]) were of parental origin. In family studies we confirmed that each parent harbours one variant. Only a maternal sample was available for testing variant inheritance for Patient 3 (SEMA7A:c.[916G>A];[=], SRA1: c.[377G>A][=]). In the mother’s DNA we identified SEMA7A variant and excluded the SRA1 variant. Thus, the latter variant was either inherited from the father or occurred de novo. In addition to the CCDC141 variant, patient 4 has two POLR3B variants (CCDC141:c.[2299G>A];[=] POLR3B:c.[1244T>C];[2570+1G>A]). Interestingly, the CCDC141 variant and splice site mutation in POLR3B were inherited from an asymptomatic mother, while the nonsynonymous POLR3B variant was of paternal origin. Patient 5 harboured paternally transmitted PROKR2 variant and two maternally transmitted variants: SPRY4 and NSMF (PROKR2:c.[889G>T];[=], SPRY4:c.[530A>G];[=], NSMF: c.[1261C>T];[=]). The patient’s mother showed no symptoms of GnRH deficiency.
Based on the literature findings, we assumed that a single heterozygous variant would not be sufficient to explain the CHH phenotype. Results from the family study strongly support the hypothesis. Thus, a synergistic role of identified variants is highly probable.
Discussion
In this study we report on new genetic findings supporting the importance of oligogenic inheritance in congenital hypogonadotropic hypogonadism. We have recognised five new oligogenic patterns/combinations: SPRY4/SEMA3A, SRA1/SEMA7A, CHD7/SEMA7A, CCDC141/POLR3B/POLR3B, and PROKR2/SPRY4/NSMF. All of the listed genes are known to contribute to phenotype of CHH, but our results point to potential new “partners” underlying digenic/trigenic inheritance. Herein we report both novel variants and those that were earlier recognised in the aetiology of CHH.
Both variants, SPRY4 c. 722C>A and SEMA3A c. 1303G>A, identified in patient 1 were previously reported in IHH/KS patients [14, 15]. SPRY4 protein acts as an inhibitor of the receptor-transduced mitogen-activated protein kinase (MAPK) and thus antagonises FGF signalling pathway [21]. Activity of this protein is important for proper regulation of neuron outgrowth, which has been proven in the animal model of Spry4–/– mice [22]. Nucleotide variation in this gene has been associated with hypogonadotropic hypogonadism with or without anosmia [15]. Semaphorin 3A is a key player in GnRH neuron migration. It has been shown in mice Sema3a-null mutants that loss of Sema3a signalling alters the targeting of vomeronasal nerves and the migration of GnRH neurons into the brain, resulting in reduced gonadal size and infertility [23]. Later it was documented that a heterozygous mutation of SEMA3A leading to haploinsufficiency, accompanied by a second genetic hit in another gene affecting the GnRH pathway, produces the KS phenotype [14, 24]. Interestingly our patient presented symptoms of CHH with normosmia. Based on the function of SEMA3A gene and its involvement in guidance of vomeronasal axons, we would have expected to observe a deteriorated sense of smell in our patient. Thus, we speculate that heterozygous missense variant of SEMA3A may not be sufficient to modify the phenotype of the olfactory system.
SEMA7A gene encodes for semaphorin 7A, a membrane-anchored guidance protein known to enhance central and peripheral axon growth. It is required for proper axon tract formation during embryonic development [25]. Only one study reported on SEMA7A gene variants (p.R148W, p.R474Q) in 2 patients. SEMA7A variants have been shown to contribute to the oligogenic phenotype of both nCHH and KS, together with KISS1R and KAL1, respectively [14]. Identified in our cohort, SEMA7A: c.618_619delCC mutation is an out-of-frame deletion and thus predicted null variant due to a premature termination codon and consequently nonsense-mediated decay. In the same subject CHD7 mutation was identified. Chromodomain helicase DNA-binding protein 7 is a transcriptional regulator, and its mutations are responsible for up to 70% of CHARGE syndrome cases. However, several heterozygous CHD7 mutations have been recognised in nCHH/KS patients with no symptoms of CHARGE phenotype. Moreover, some variants were recognised in patients presenting with various phenotypes of nCHH, KS, or CHARGE syndrome [26–28]. Thus, a growing amount of evidence supports the hypothesis that nCHH/KS could be a milder allelic variant of CHARGE syndrome. Interestingly, in the patient’s family a case of choanal atresia was present. Association of Kallmann syndrome and choanal atresia was previously reported in a paper by Klein et al. [29]. The authors described several cases of both conditions affecting members of one family. Because the inheritance did not exactly match an autosomal dominant pattern it was called “irregular AD mode of inheritance”. We believe that what was observed and reported is now acknowledged as oligogenic inheritance, a phenomenon not known 30 years ago. Missense mutation CHD7 c.2840G>A revealed in our patient predicts a substitution Arg947Gln at the protein level and was previously reported in a single case of CHARGE syndrome [19]. Due to the lack of sufficient evidence the variant was previously classified as probably benign. In this case, reporting a second unrelated patient harbouring the same mutation seems to be a sufficient argument to revise variant and assort it from class 2 to class 3 (VUS). Our finding also provides further support to the allelic relationship of nCHH/KS, choanal atresia, and CHARGE syndromes.
SRA1 is a transcriptional coactivator of steroid nuclear receptors. To date, four missense mutations in the SRA1 gene have been reported in CHH: p.P20L, p.Q32E, p.T103A, and p.I79T [30]. Mutations were revealed in families presenting homozygous, compound heterozygous, and digenic pattern of inheritance. In the digenic case, SRA1 mutation was accompanied by PNPLA6 missense variant. Herein we report a patient harbouring heterozygous SRA1 mutation c.377G>A that predicts substitution of arginine for histidine at residue 126 (p.Arg126His) and SEMA7A gene mutation c.916G>A (p.Val306Ile). Heterozygous SEMA7A mutations were noted in both normosmic HH and KS, but in the case of anosmia the second reported variant was a deleterious mutation of KAL1 gene, which is crucial for the development of both GnRH and olfactory neurons. Therefore, SRA1 and SEMA7A seem to have no impact on olfactory function, which is concordant with the phenotype of our patient.
CCDC141 encoding for coiled-coil domain containing protein was recently shown to play an important role in GnRH neuron migration [31]. Its heterozygous mutations were identified in patients with Kallmann syndrome and normosmic CHH [32, 33]. Herein we report a patient with a novel variant of CCDC141, a missense mutation (c.2299G>A) that leads to Asp to Asn substitution at residue 767. The variant was predicted as deleterious by several programs used for in silico analysis. In the same patient we have also identified two biallelic mutations in POLR3B gene, which encodes for the polymerase III subunit. The T-to-C transition c.1244T>C leading to an amino acid change p.(Met415Thr) was previously reported in IHH and 4H leukodystrophy [20, 34, 35]. The mutation was identified in multiple individuals, but all of them were carrying a second missense mutation in the POLR3B gene. These observations led to the question of whether the p.(Met415Thr) variant is harmful at all, although prediction programs are congruent in terms of variant pathogenicity. Alternatively, a combination of two mutations impacting both alleles is necessary to evoke the effect [20]. In addition to the p.(Met415Thr) variant, we have identified an intronic mutation c.2570+1G>A in POLR3B. According to NNSPLICE, the mutation creates a splice defect. Notably, the proband’s parents were heterozygous for POLR3B mutations, the splice site variant was inherited from the mother, while the missense POLR3B variant was of paternal origin. Thus, our finding supports the hypothesis that a severe effect on phenotype can be observed when the Met415The variant is combined with a mutation affecting the transcription from the second allele.
Patient 5 carries a heterozygous mutation in SPRY4 c.530A>G (p. Lys177Arg), which was implicated in KS [15]; moreover, he harbours two novel missense mutations c.889G>T (p.Val297Ile) in PROKR2 and c.1261C>T (p.Leu241Phe) in NSMF gene. Notably, the SPRY4 and NSMF variants were of maternal origin, while the PROKR2 variant was paternal. PPROKR2 coding for prokineticin receptor 2 is a gene implicated in both IHH and KS via defective GnRH neuron development [9]. NMDA receptor synaptonuclear signalling and neuronal migration factor encoded by NSMF gene, previously known as NELF, has been shown to contribute to CHH pathogenesis [36].
Herein we present several sequence variants in nCHH and KS patients. According to the ACMG–AMP criteria [37, 38], we classified them as variants of uncertain significance (VUS, class 3), probably pathogenic (class 4) or pathogenic (class 5). Oligogenic inheritance does not show regular Mendelian segregation patterns, nor does it follow Mendelian mechanisms. However, in the absence of adequate tailored criteria for variant classification we used the one meant for interpretation of sequence variants in monogenic diseases. We thus believe that the variant categories assigned in our study might be underestimated.
In conclusion, with accumulating data, it is becoming increasingly clear that oligogenicity may account for the phenotype in many complex diseases. Hopefully, with the growing evidence of oligogenic patterns, it will be possible to fine-tune guidelines and provide a tool for precise sequence variant classification in non-Mendelian conditions.