Introduction
Obstructive sleep apnoea (OSA) is a form of sleep-disordered breathing that is caused by repetitive closure of the upper airway during sleep, resulting in discrete episodes of obstructive apnoeas, hypopnoeas, or respiratory effort-related arousals. The prevalence of OSA, defined as an apnoea-hypopnoea index (AHI) ≥ 15 or an AHI ≥ 5 with excessive daytime sleepiness, is about 15% in men and 5% in women [1].
However, the prevalence of OSA in the obese population is very high, with an estimated 60.5% of obese patients with metabolic syndrome having moderate to severe OSA (AHI ≥ 15) [2]. It is equally prevalent in obese patients with type 2 diabetes mellitus. A study done by Foster et al. has shown that over 86% of such patients had mild OSA (AHI ≥ 5), 30.5% had moderate OSA (15 ≤ AHI < 30), and 22.6% had severe OSA (AHI ≥ 30) [3].
Furthermore, a recently published study revealed that 85.7% of morbidly obese patients who underwent bariatric surgery had OSA, defined as a respiratory disturbance index (RDI) of 5 or more. Of these patients, the vast majority (75.9%) had severe OSA [4].
It is well known that OSA is the main cause of chronic intermittent hypoxia (CIH). Research on the impact of CIH on cardiovascular complications began more than 25 years ago when researchers found that inducing cyclic hypoxia caused daytime hypertension [5]. Since then, there has been an increase in basic and clinical studies on animal models and humans. Chami et al. studied a group of 2721 participants who underwent two in-home polysomnograms 5 years apart. The mean change in AHI was greater in subjects who had a cardiovascular event compared to those who did not (5.86 vs. 2.67) [6].
In this article, we will review the pathophysiology of OSA as an independent risk factor for cardiovascular disease and discuss the most common markers that have been studied in animal and human models. We will also go over the impact of OSA management on the reversibility of atherosclerosis. We searched in PubMed, Medline, Embase, and Scopus using the terms “OSA and cardiovascular”, “OSA and atherosclerosis”, “OSA and diabetes mellitus”, “OSA and hypertension”, “OSA and dyslipidemia”, and “OSA and hyperlipidemia”. We reviewed the effect and management outcomes of OSA on each condition.
Chronic intermittent hypoxia and the pathophysiology of atherosclerosis
Several chronic inflammatory markers have become well established as risk factors for atherosclerosis, including, but not limited to, reactive oxygen species (ROS) [7], hypoxaemia-induced factor 1α (HIF-1α) [8, 9], nuclear factor kappa B (NF-κB) [10], matrix metallopeptidase-2 (MMP-2) [11], pentraxin-3 [12], and nesfatin-1 [13] (Figure 1).
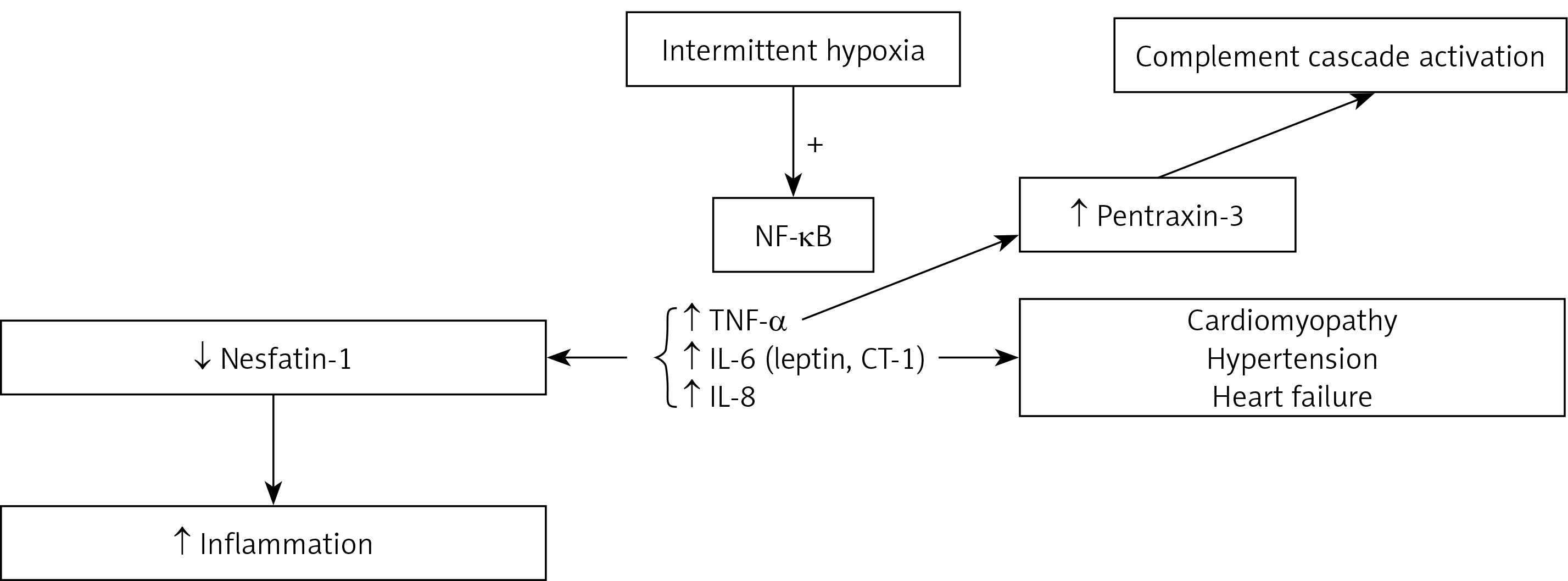
Chronic intermittent hypoxia is a prominent feature of OSA, and studies have shown that CIH plays a significant role in inducing ROS, reactive nitrogen species, and oxidative stresses that initiate and propagate inflammatory responses. This was demonstrated in a study of well-described OSA rat models induced with simultaneous intermittent hypoxia, where the rats were exposed to intermittent hypoxia 8 h daily for 5 weeks. Levels of HIF-1α, NF-κB, interleukin 6 (IL-6), and MMP-2 were significantly increased in rats with CIH compared to controls [14].
Nuclear factor κB is a family of transcription factors composed of members of the Rel family. In the nucleus, NF-κB potentiates the inflammatory cascade by upregulating cytokines such as tumour necrosis factor α (TNF-α), IL-6, and interleukin 8 (IL-8) [15]. Hence, inhibiting NF-κB activation might be a future therapeutic option.
Leptin and cardiotrophin-1 (CT-1) are members of the IL-6 super family. They are considered as risk factors for cardiovascular diseases. Specifically, CT-1 induces hypertrophy of cardiac myocytes in vitro and contributes to cardiomyopathies and heart failure [16]. Levels of both markers are significantly increased in patients with OSA compared to controls, and are significantly elevated in severe OSA compared to mild to moderate OSA [16].
Besides the elevation of common biomarkers such as C-reactive protein, a novel biomarker called pentraxin-3 is also elevated in the OSA population. Pentraxin-3 is produced by neutrophils, macrophages, smooth muscle, and endothelial cells in response to interleukin 1 (IL-1) or TNF-α signaling. Pentraxin-3 has been identified as an initiator of the complement cascade [17, 18].
Nesfatin-1 is another biomarker that has an anti-inflammatory effect, initially discovered for its role in central hypothalamic appetite suppression and a potential satiety regulator. It was subsequently detected in adipocytes [19]. Although nesfatin-1 is positively correlated with serum IL-6, IL-8, and TNF-α in chronic obstructive pulmonary disease (COPD) patients [20], Shen et al. examined nesfatin-1 levels in OSA patients and found them to be significantly reduced in comparison to weight-matched controls without OSA [21].
Fibrinogen is another biomarker that has been shown to predict cardiovascular events, implying an inflammatory role [22]. Shamsuzzaman et al. found, in a study of 36 OSA patients and 18 controls, that fibrinogen levels were significantly elevated in patients with severe OSA compared to both controls (p = 0.003) and subjects with mild OSA (p = 0.02) [23].
Drager et al. performed an experimental study on mice, which showed that CIH increased adipose angiopoietin-like 4 (an encoded protein that regulates lipid metabolism), decreased adipose lipoprotein lipase, increased fasting levels of plasma triglycerides (TG) and very low-density lipoprotein cholesterol, and increased the size of atherosclerotic plaques. The investigators also recruited obese patients who underwent bariatric surgery, where they found significant correlation between angiopoietin-like 4 mRNA levels in subcutaneous adipose tissue, the severity of OSA, and nocturnal oxyhaemoglobin desaturation (p < 0.05) but without significant correlation with their body mass index (BMI) [24].
Chronic intermittent hypoxia, nitric oxide bioavailability, and endothelial dysfunction
The endothelium plays a significant role in the stability of vascular tone and structure. Studies have shown that endothelial dysfunction is a risk factor for atherosclerosis. Hoffmann et al. looked at the effect of serum factors on endothelial cell migration, an important mechanism of endothelial repair, and found that cells incubated in serum from patients with OSA showed blunted migration compared to healthy patients [25]. Therefore, elevated markers of endothelial dysfunction seen in OSA can be used as surrogates to measure the degree of endothelial disruption [26].
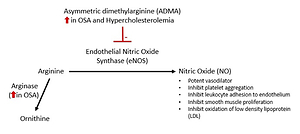
Nitric oxide (NO) is produced from arginine via endothelial nitric oxide synthase (eNOS) [27]. NO acts as a potent vasodilator by increasing the production of cyclic guanosine monophosphate (Figure 2). In addition to vasodilation, NO inhibits platelet aggregation, adhesion of monocytes and leukocytes to the endothelium, smooth muscle cell proliferation, and oxidation of low-density lipoprotein (LDL) [28–31]. Therefore, NO is a protective factor against atherosclerosis. Inhibiting NO causes endothelial dysfunction and promotes progression of atherosclerosis.
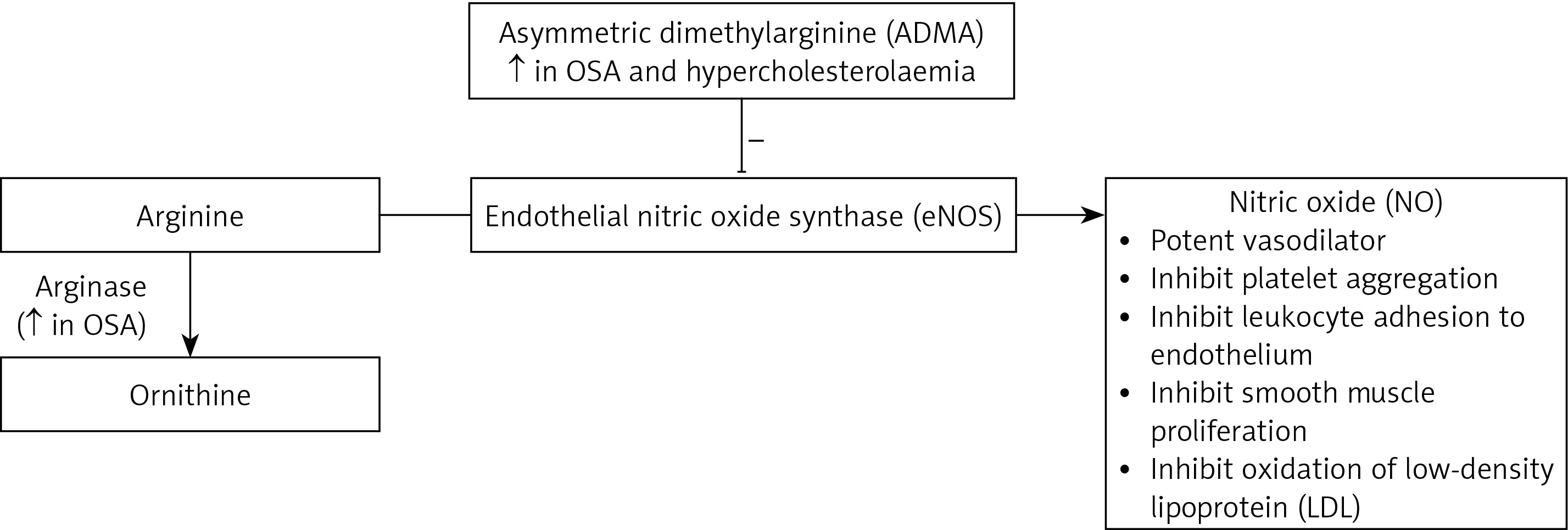
Asymmetric dimethylarginine (ADMA) is an endogenous inhibitor of eNOS that is also produced in the endothelium. Studies have shown that plasma ADMA levels are elevated in renal failure and hypercholesterolaemia [32, 33].
Arginase is an enzyme that is involved in the inflammatory and immune response by competing with eNOS, which, in turn, results in an imbalance in arginase/eNOS regulation and therefore a reduction in NO bioavailability [34] (Figure 2).
In animal models, validated rodents were intermittently exposed to reduced oxygen (from 21% to 6–7%) over 30-second periods for 8 weeks. The basal production of NO was significantly lower (AUC: 65.0 ±3.2) and the plasma levels of ADMA were higher (AUC: 0.63 ±0.04) in rodents exposed to intermittent hypoxaemia [35].
Studies have also shown elevated ADMA plasma levels in OSA patients. In a study of 64 subjects, Barceló et al. measured the plasma ADMA in 23 OSA patients with concurrent cardiovascular risk factors and 18 subjects without risk factors and found it to be significantly higher (1.17 ±0.13 μmol/l) compared to controls (0.87 ±0.07 μmol/l, p < 0.01) [36]. However, this study found no elevation in plasma endothelin-1 (ET-1) levels, which contradicted other studies [37, 38].
Interestingly, Ozkan et al. found that serum levels of NO in OSA subjects were significantly lower than those of controls (p < 0.05) while plasma ADMA levels did not reach a statistically significant difference [39].
Endothelin-1 is another potent vasoconstrictor that activates endothelin receptors type A and B in vascular smooth muscle and endothelial cells [40]. Gjørup et al. studied the levels of multiple factors including ET-1 and angiotensin II in 32 patients with OSA and 19 healthy controls. Nocturnal plasma levels of ET-1 were significantly higher in moderate to severe OSA subjects compared to controls [41]. No difference was found in angiotensin II levels.
Rho-associated kinases (ROCKs) are protein kinases that are characterised by their ability to mediate the formation of RhoA-induced stress fibres and focal adhesions. ROCKs play significant roles in cellular apoptosis, growth, metabolism, and migration by controlling the actin cytoskeletal assembly and cell contraction. Studies have shown that ROCK activation was associated with atherosclerosis and arterial hypertension in rat models [42] and humans [43]. Interestingly, Li et al. noticed a correlation between induced CIH and ROCK pathway upregulation [44].
The impact of obstructive sleep apnoea on cardiovascular risk factors
Obstructive sleep apnoea is associated with increased coronary artery disease risk and multiple cardiac disorders such as cardiomyopathy, arrhythmia, systolic and diastolic dysfunction, and heart failure, as well as an increased risk of stroke [45]. Specifically, in patients with moderate to severe OSA [46]. The association between OSA and cardiovascular disease (CVD) is well supported by both animal and human studies (Tables I and II).
Table I
Evidence from non-human studies linking obstructive sleep apnoea and cardiovascular disease
Table II
Evidence from human studies linking obstructive sleep apnoea and cardiovascular disease
Obstructive sleep apnoea and carotid intima-media thickness
Carotid intima-media thickness (CIMT) is a well-known indicator of atherosclerosis. A study of 80 patients with OSA and 40 controls has shown that CIMT in patients with moderate-severe OSA (0.95 ±0.09 mm) was significantly increased when compared to those with mild OSA (0.76 ±0.1 mm; p < 0.01) and controls (0.65 ±0.1 mm; p < 0.01) [47].
Macrophages are important contributors to the genesis of atherosclerotic plaque. An in vitro study on murine atherosclerotic plaques showed that macrophages incubated in 1% oxygen showed an increased triglyceride content of 220% relative to normoxic cells (p < 0.01) [48].
A post-hoc analysis done by Liao et al. showed that patients with severe OSA had significantly higher mean CIMT than healthy controls and patients with mild-moderate OSA (p < 0.01). However, their multivariate linear regression could not prove that OSA was an independent factor for increased CIMT [49]. In contrast, a meta-analysis done by Zhou et al. adjusted for major confounders such as age, gender, blood pressure, cholesterol, and glucose found that OSA was an independent risk factor for carotid atherosclerosis. In addition, they also found a dose-dependent relationship between AHI and CIMT [50].
A study of 180 non-apnoeic snorers and non-snorers, based on full-night home-based polysomnography, showed that mean CIMT increased with increased snoring time throughout the night in women but not in men [51]. Therefore, because studies have shown a direct correlation between CIH and biomarkers, snoring could be an additional minor contributor in women. However, further studies are needed in this regard.
Interestingly and controversially, a recently published multivariable linear regression analysis showed that the presence or absence of OSA significantly contributed to the peak cardiac troponin I level, which was 54% lower in patients with OSA than in those without OSA (defined in this study as AHI < 15). Suggesting that patients with OSA may experience less severe myocardial injury [52]. However, Belaidi et al. have shown, in animal models, that CIH in patients with OSA resulted in an increase in infarct size [53].
Furthermore, in a study of 100 patients with an ST-elevation myocardial infarction, who underwent primary percutaneous coronary intervention (PCI) within 12 h of onset, Nakashima et al. reported that the incidence of systolic retrograde flow (a sensitive echocardiographic index of severely impaired myocardial tissue perfusion) was higher in OSA patients than in the controls (6% vs. 31%, p = 0.005). Similar outcomes were found with ST-segment resolution (STR), a pre-specified electrocardiographic index of impaired tissue perfusion, where the incidence of STR < 50% was higher in OSA patients than in controls (31% vs. 60%, p = 0.003), suggesting that OSA may induce more severe microvascular injury related to the ischaemia–reperfusion process [54]. Additionally, a few meta-analyses found that patients with OSA had a significantly increased risk of recurrent cardiovascular event after PCI, cardiac death, and coronary revascularisation [55–57].
Obstructive sleep apnoea and hypertension
Over 50% of individuals with OSA have hypertension [58]. In a study of 125 patients with resistant hypertension, OSA (AHI > 15) was present in 64.0% of the patients [59]. Another study showed that nocturnal blood pressure variability was significantly greater in patients with severe OSA (AHI > 29) than in patients with milder OSA [60]. Additionally, OSA was found to be significantly associated with both essential and resistant hypertension, with a trend of higher risk with more serious OSA [61].
The aetiology of hypertension in OSA has been linked to the increase in sympathetic activity as a result of CIH and/or apnoea. Fletcher et al. measured urinary epinephrine, norepinephrine, metanephrine, and normetanephrine in 8 patients with severe apnoea before and after they underwent surgical placement of tracheostomies as curative therapy for their apnoea [62]. The results were compared with samples collected from 5 obese hypertensive subjects without apnoea. Although urinary epinephrine and metanephrine were not different between subjects and controls, urinary norepinephrine and normetanephrine were significantly higher in apnoeic subjects pre-tracheostomy compared with either controls or their own values post-tracheostomy (p < 0.01). Diurnal variation was not seen before tracheostomy reflecting the increased nocturnal sympathetic activity. The elevation of daytime catecholamines with return to control levels post-tracheostomy suggests increased sympathetic activity throughout the day.
In another study published in 1995, serum and 24-hour urinary norepinephrine were measured in 43 subjects (including hypertensive and normotensive individuals, with and without OSA) consuming similar diets [63]. 24-hour urinary norepinephrine levels were significantly higher in apnoeics (58.2 vs. 40.2 ng in non-apnoeics, p < 0.002) during both day and night. However, plasma norepinephrine levels were not significantly elevated in apnoeic patients but were elevated in hypertensive patients both during sleep and in the morning (p < 0.05).
A study done by Ziegler et al. monitored plasma norepinephrine (NE) levels, clearance, and release rate among 65 subjects who breathed room air, a hypoxic gas mixture, and the hypoxic mixture combined with intermittent breath holding [64]. All individuals underwent two-night polysomnography (apnoeics were defined as having RDI ≥ 20). The apnoeics’ mean plasma NE level was higher compared with the non-apnoeics (p = 0.017). The NE-release rate response to hypoxia and breath holding differed between hypertensives and normotensives (p < 0.001) and between apnoeics and non-apnoeics (p < 0.001). Normotensive apnoeics had the greatest increase in NE release during hypoxia, and hypertensive apnoeics maintained their increased sympathetic nervous release of NE in the daytime.
A systematic review of 24 studies showed that most studies reported an elevation of norepinephrine and muscle sympathetic nerve activity, both during sleep and wakefulness, among individuals with sleep apnoea. Epinephrine increase was less frequent. However, the studies were rarely controlled for well-known confounders of sympathetic activity, making the validity of the findings questionable [65].
Obstructive sleep apnoea and insulin resistance
The multicentre Sleep Heart Health Study (SHHS), which had a cohort of 2565 participants, allowed Punjabi et al. to examine the relationship between OSA and glucose intolerance. They found that relative to subjects with an RDI < 5.0, subjects with mild OSA (RDI = 5–14.9) and moderate to severe OSA (RDI ≥ 15) have adjusted odds ratios of 1.27 (95% CI: 0.98–1.64) and 1.46 (95% CI: 1.09–1.97), respectively, for fasting glucose intolerance (p < 0.01). Furthermore, hypoxaemia (average SpO2 below 90%) during sleep was independently associated with glucose intolerance, based on either fasting glucose values or 2-hour glucose values [66].
Using the homeostasis model assessment (HOMA) index as a surrogate of insulin resistance, the investigators of SHHS found that subjects with moderate to severe OSA (RDI ≥ 15) have higher values of the HOMA index, indicating higher insulin resistant states, regardless of their age, gender, ethnicity, smoking status, BMI, waist circumference, or sleep duration.
A smaller study of 30 obese subjects with OSA, 27 weight-matched controls, and 20 normal subjects showed that the insulin sensitivity index (whole body and hepatic) was significantly lower in patients with OSA even after adjusting for age, BMI, and waist-to-hip ratio [67]. Similar outcomes have been reported in other studies [61, 68–71].
Interestingly, OSA also seems to contribute to the development of neuropathy in diabetic patients. A meta-analysis done by Gu et al. consisting of 11 studies found a significant correlation between OSA and development of diabetic neuropathy in type 1 diabetics [72], supporting the idea that OSA also aggravates nerve damage in addition to its association with insulin resistance.
Obstructive sleep apnoea and dyslipidaemia
In a study of 152 subjects with metabolic syndrome, OSA was independently associated with significantly higher levels of TG, LDL cholesterol, cholesterol/high-density lipoprotein (HDL) ratio, and TG/HDL ratio [2, 73]. In a meta-analysis of 11 studies, the risk of developing metabolic syndrome in patients with OSA was estimated to be 1.72 times higher than in the overall population [74].
An experimental study on rodents exposed to intermittent hypoxia for just 5 days showed that an increase in fasting serum levels of total cholesterol (TC), HDL cholesterol, phospholipid, TG levels, and liver TG content occurred in lean mice but not in leptin-deficient obese mice [75].
In humans, the available data have been conflicting so far. In a cross-sectional study of 2983 subjects recruited from the Shanghai Sleep Health Study, multivariable logistic regression analyses demonstrated that only LDL cholesterol was independently associated with OSA [76]. A large-scale meta-analysis by Nadeem et al., which included 64 studies, found increased TC, LDL, HDL, and TG in subjects with OSA compared to healthy controls [73]. Additionally, they found that the degree of dyslipidaemia (HDL and TG) is correlated with severity of OSA. On the other hand, a study of 200 participants demonstrated statistically significant higher levels of TG and TC and lower levels of HDL cholesterol in OSA positive patients compared to OSA negative controls with BMI ≤ 30 kg/m2, but there was no significant difference in patients with BMI > 30 [77].
The SHHS showed that HDL cholesterol levels were inversely related to RDI levels in women and in men under 65 years of age, but not in men aged 65 years or more [78].
The impact of obstructive sleep apnoea management on atherosclerosis risk factors
The impact of obstructive sleep apnoea management on atherosclerosis
Several studies have looked at the impact of continuous positive airway pressure (CPAP) treatment on the reversibility of OSA-related atherogenic factors. Available data for the use of CPAP so far have been conflicting [79]. Significant reductions in major cardiovascular events have been reported, but only seen in observational studies, not in randomised controlled trials [79–81]. A meta-analysis by Aslan et al. found that CPAP treatment significantly increased left ventricular ejection fraction (LVEF) but found no reduction in all-cause mortality or occurrence of cardiovascular events [82].
A group of 90 matched patients diagnosed with OSA were blindly randomised to therapeutic vs. subtherapeutic CPAP for 3 months. A variety of parameters such as fat thickness, CIMT, and adipokines were measured at baseline and at 3 months in both groups. While short-term therapeutic CPAP did not significantly reduce visceral fat thickness or CIMT, it did significantly reduce adiponectin (a hormone secreted by adipocytes with both anti-atherogenic and anti-inflammatory properties) and increase irisin (a hormone produced by skeletal muscles, elevated levels of which have been shown to be associated with a lower risk of insulin resistance) [83].
Other studies have shown an increase in adiponectin with CPAP therapy [84]. However, a recent meta-analysis suggested that CPAP therapy has no impact on adiponectin in OSA patients [85]. In summary, available data in this area remains contradictory.
Impact of obstructive sleep apnoea management on diabetes mellitus and hypertension
In a trial of 64 diabetic patients with moderate to severe OSA (AHI ≥ 15), who were randomised to either CPAP or no treatment (control), treatment with CPAP for 3 months did not improve glycaemic control defined by glycosylated haemoglobin (HbA1c) or microalbuminuria, but it did lower both systolic and diastolic blood pressures significantly [86]. A meta-analysis by Sun et al. also found that CPAP treatment significant reduced both systolic and diastolic BP levels, and 24-hour BP levels [87]. They also reported a significant decrease in risk of cardiovascular events at 28 months follow-up compared to the control group.
Another multi-centre study of 298 participants with type 2 diabetes and an oxygen desaturation index ≥ 15 events per hour showed no change in HbA1c after 6 months of CPAP treatment versus no intervention [88]. However, it did demonstrate a greater fall in diastolic blood pressure over 6 months in the CPAP therapy adherent group than in the usual care group after adjustment for BMI (–4.8 vs. –1.0 mm Hg; p = 0.002) but had no effect on systolic blood pressure. A meta-analysis also found no improvement in HbA1C or fasting glucose level after more than 12 weeks of CPAP therapy [89].
In a recent study, treatment of hypertensive patients, some of who had OSA, with losartan 50 mg daily for 6 weeks was followed by either add-on CPAP treatment or no CPAP. Losartan resulted in a significant reduction in blood pressure in both groups, but the reduction was less in patients without OSA. Adherence to CPAP treatment for at least 4 h per night showed significant reduction in all 24-hour blood pressure values, while use of CPAP, less efficiently, only reduced night-time systolic blood pressure [90].
A group of 20 obese patients with type 2 diabetes mellitus and newly diagnosed moderate to severe OSA underwent continuous glucose-monitoring (using a system that measured interstitial glucose every 5 min) during polysomnography and after an average of 41 days of CPAP treatment. Even though the study was limited by the lack of a control group, a statistically significant reduction in the mean glucose level during sleep with greater stabilisation was noted, but there was no change in their HbA1c [91].
Longer CPAP treatment (12–52 weeks) has shown significant reduction in HbA1c and better glucose control in CPAP-adherent participants with poorly controlled diabetes mellitus [92, 93].
In a meta-analysis of 20 studies of 2713 patients who underwent sleeve gastrectomy (SG), 1626 patients reached the minimum 5-year follow-up period (ranging from 5 to 11 years). Their mean preoperative BMI was 46.9 kg/m2. Resolution or improvement of type 2 diabetes, arterial hypertension, and dyslipidaemia occurred in 77.8%, 68.0%, and 65.9% of patients, respectively. The 8 studies that investigated the effect of SG on OSA revealed a 75.8% resolution or improvement [94]. Though this meta-analysis did not examine cardiovascular outcomes directly, it did investigate major risk factors for atherosclerosis.
The impact of obstructive sleep apnoea management on the hypoxia/reoxygenation process
In moderate to severe OSA intermittent hypoxia results in ROS production, potentially inducing HIF-1α. This, in turn, promotes the transcription of numerous adaptive genes, some of which are deleterious to the cardiovascular system, such as endothelin-1 [95, 96]. Additionally, the process of hypoxia/reoxygenation results in mitochondrial dysfunction causing an increase in ROS production and subsequent vascular damage.
NADPH oxidases (NOX) are enzymes that produce ROS. Isoforms of NOX are localised within circulating leukocytes. However, some components are present in the vascular system within endothelial and smooth muscle cells [97, 98].
In a murine model of induced sleep apnoea in which mice were exposed to CIH for 6 weeks, Schulz et al. investigated the role of apocynin (a NOX inhibitor) on the prevention and treatment of hypertension. Blood pressure was monitored by an implanted radio telemetry system in the abdominal aorta. CIH mice received intraperitoneal injections of apocynin (30 mg/kg/day) or placebo. Compared to those not exposed, wild type mice exposed to CIH showed a significant blood pressure elevation after 2 weeks of exposure, and it progressively increased for the duration of the experiment.
Wild type mice that were treated by apocynin did not show arterial hypertension in response to CIH, unlike those treated with placebo. In addition, NADPH oxidase-2 knockout mice subjected to CIH were protected from the development of arterial hypertension [99].
Nuclear factor of activated T-cells 3 isoform (NFATC-3) is a protein-coding gene that plays a role in vascular smooth muscle differentiation, contractility, and hypertrophy [100–103]. The nuclear accumulation of NFATC-3 in vascular smooth muscle is increased by Gq-coupled receptor agonists such as uridine triphosphate, angiotensin II, and endothelin-1. This results in vascular remodelling. de Frutos et al. demonstrated in mice that the endothelin receptor antagonist bosentan could prevent CIH-induced arterial remodelling and hypertension [104].
The impact of obstructive sleep apnoea management on carotid intima-media thickness
A recently published meta-analysis by Chen et al. of 7 studies with a total of 167 cases showed no significant change of CIMT before and after CPAP treatment in OSA patients [105]. However, a subgroup analysis demonstrated that CIMT was significantly decreased after CPAP treatment in more severe OSA patients with AHI ≥ 50 (95% CI: 0.022–0.124, p = 0.005) and patients with therapeutic duration of at least 6 months (95% CI: 0.019–0.223, p = 0.021). A separate study by Ng et al. also found no significant reduction of CIMT with CPAP treatment over 3 months in patients who received therapeutic CPAP (> 4 cm H2O) or subtherapeutic CPAP (3–4 cm H2O) [106].
This contrasts with an earlier prospective study done by Hui et al., in which they looked at CIMT reduction with CPAP treatment over the course of 1 year compared to patients who received standard treatments [107]. They found that patients treated with CPAP had a significant reduction in CIMT when measured at 6 months and 12 months, with average CPAP usage of 4.6 and 4.7 h per night, respectively. This effect was seen in both patients with and without existing cardiovascular disease [107].
Impact of obstructive sleep apnoea management on nitric oxide bioavailability and endothelial function
Different indicators have been used to indirectly assess endothelial function, including flow-mediated dilatation (FMD), venous occlusion plethysmography, peripheral arterial tonometry (PAT), NO, and bronchial artery diameter.
In a systematic review of 8 randomised controlled trials (totalling 245 OSA patients) published between 2004 and 2013 looking at the impact of CPAP on endothelial function using FMD and other validated indicators, Schwarz et al. found that therapeutic CPAP significantly improved endothelial dysfunction in 6 of the 8 reviewed studies [108]. The same researchers also did a meta-analysis on 4 randomised controlled trials (totalling 150 patients) in which they found that CPAP therapy (ranging from 2–24 weeks) increased the FMD by 3.87% (95% CI: 1.93–5.80 with p < 0.001) compared to controls.
A prospective study of 84 patients with OSA (AHI > 10) revealed that 4–12 weeks of CPAP treatment significantly improved FMD. Interestingly, after 1 week of CPAP withdrawal, the brachial artery diameter dropped significantly compared with the week-12 visit, but the reduction in FMD was not statistically significant [109].
A study was done in France on 150 patients with severe OSA (AHI of 30 or more), who were using mandibular advancement devices (MAD) due to CPAP intolerance. After 2 months of effective MAD use (after a period for MAD optimisation), Gagnadoux et al. showed no significant change in the PAT [110]. However, prior studies on MAD have shown significant improvement in PAT [111] and equivalent response when MAD was compared to CPAP treatment in patients with OSA [112].
Because studies have shown that patients with OSA have higher levels of microcirculatory peroxynitrite (an oxidant and nitrating agent) deposits [113] and imbalanced arginase-1 to eNOS expressions [114], Krause et al. investigated the benefits of using arginase inhibitor 2(S)-amino-6-boronohexanoic (ABH) and the antioxidant N-acetylcysteine (NAC) on CIH-induced hypertensive rats to determine if this combination or either of them individually has any impact on hypertension and/or endothelial dysfunction [115]. While the rats were exposed to CIH for total of 35 days, the researchers started 2(S)-amino-6-boronohexanoic acid (ABH), N-acetylcysteine (NAC), or the combination (ABH/NAC) from day 14 until day 35. The researchers showed that CIH-induced hypertension was reverted by ABH, NAC, and the combination. However, only the combination of ABH and NAC reverted the vascular reactivity and oxidative stress in the CIH-induced rats.
Furthermore, fasudil, a ROCK inhibitor, was studied by Li et al. in rat models with induced CIH [44]. They showed that fasudil treatment reduced endothelial dysfunction markers significantly (improved vascular response, increased NO levels in serum and aortic tissues, decreased ET-1 in serum and aortic tissue, and decreased the expression of RhoA and ROCK-2 protein) in the CIH group.
Discussion
Obstructive sleep apnoea is a highly prevalent disease in the obese population, especially in diabetic and hypertensive subjects. Moderate to severe OSA is a well-known cause of CIH, which in turn is considered to be a contributor to atherosclerosis. Despite the existence of many studies that have demonstrated the link between OSA and atherosclerosis, other studies have not clearly shown that OSA treatment is a significant risk factor modification for atherosclerosis. However, several other studies have demonstrated that reversing CIH does appear to be protective. Despite these controversial data, it seems that there is a trend towards the benefits of OSA treatment on CVD. We believe that the future is promising despite the need for larger prospective clinical and long-term studies to determine the impact of OSA treatment on atherosclerosis and to help define future guidelines.