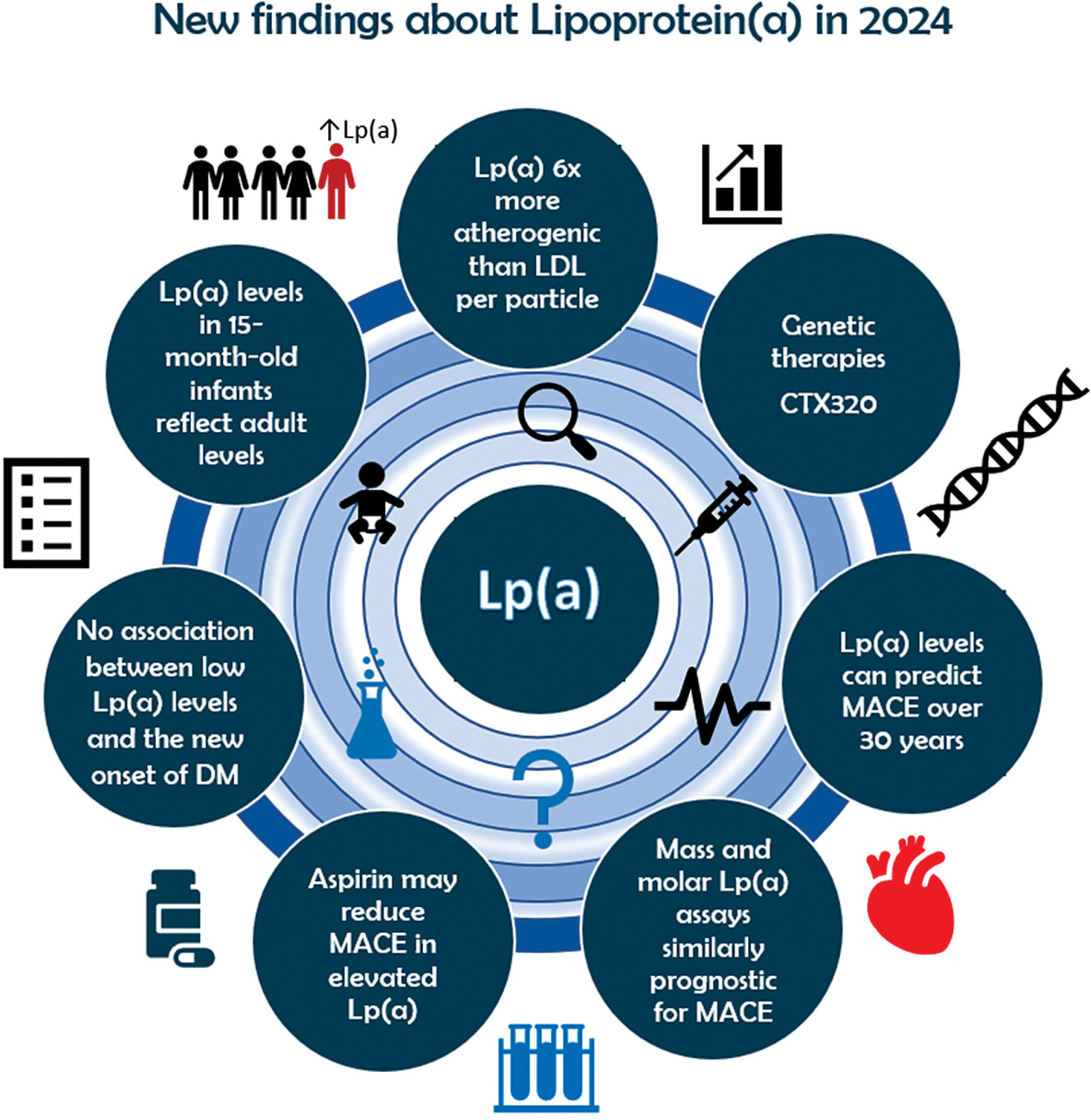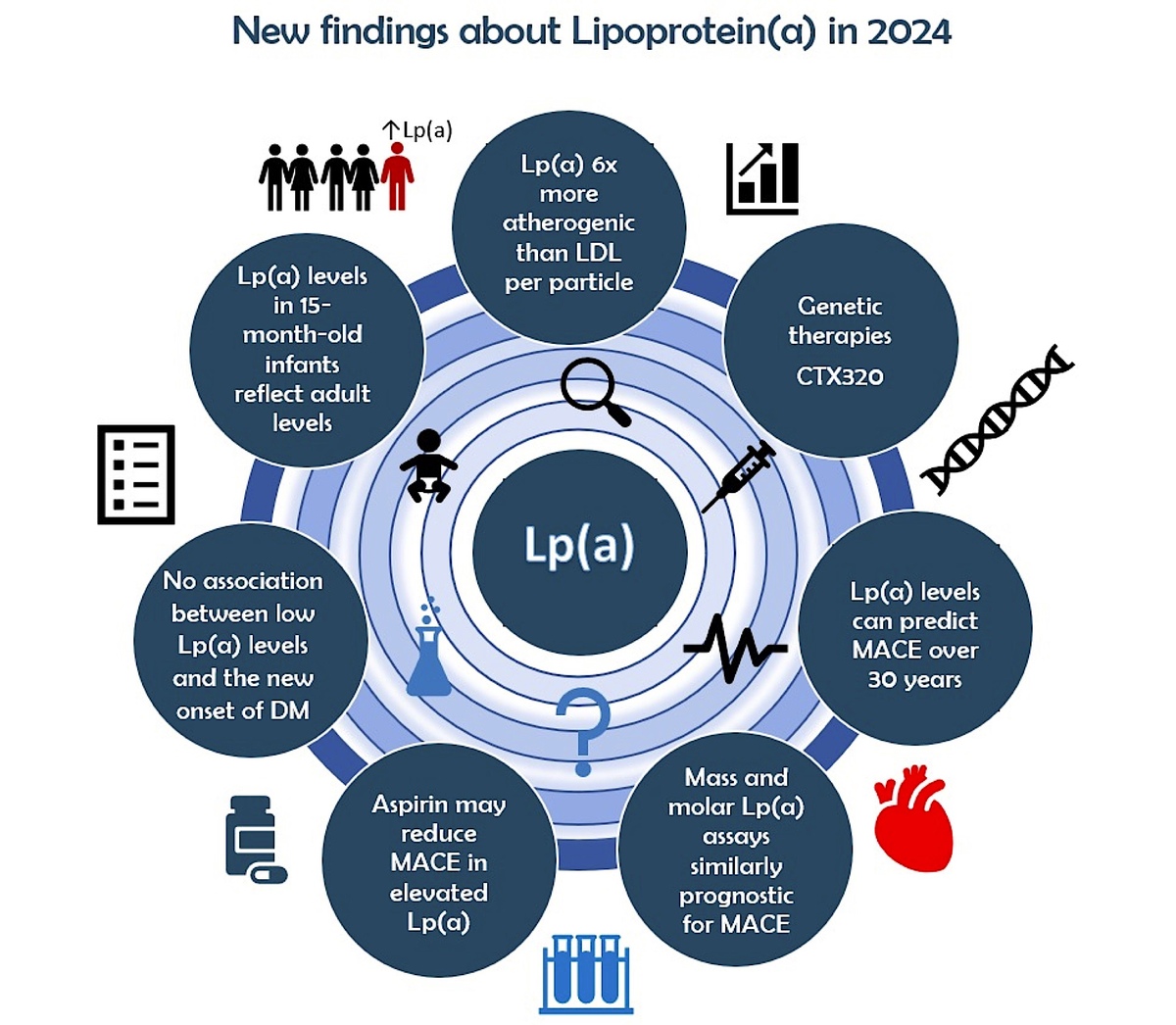
Introduction
Elevated lipoprotein(a) [Lp(a)] accounts for a significant component of residual cardiovascular disease risk (CVD) [1]. Globally, elevated Lp(a) levels (≥ 50 mg/dl or ≥ 125 nmol/l) are estimated to affect over 1.5 billion people [2]. The contribution of Lp(a) to cardiovascular risk varies, ranging from 13.5% in relatively healthy patients to 20% in patients at high and very high CVD risk, which suggests that Lp(a) may contribute to the baseline risk [3–5]. Individuals with the highest Lp(a) levels face as much as a 31% increased risk of CVD and a 42% greater likelihood of experiencing an atherosclerotic cardiovascular disease (ASCVD) event [6]. Moreover, Lp(a) concentrations exceeding 180 mg/dl are associated with a cardiovascular risk comparable to that in patients with heterozygous familial hypercholesterolemia (heFH) [7].
Apolipoprotein B100 (apoB), which is present as a single copy per particle in both Lp(a) and low-density lipoprotein (LDL), has been used to directly compare the relative atherogenicity of these two kinds of lipoproteins [1]. In the UK Biobank cohort, it was found that the Lp(a) particle has a more than 6-fold stronger association with CVD risk than the LDL particle [8]. The results of this genetic analysis were similar to the epidemiological study of Marston et al. [9]. It is important to note, however, that in most people LDL particles are present in greater amounts than Lp(a) particles; thus, the overall CVD risk associated with LDL is higher [10]. Another important issue is that exposure to high levels of Lp(a) starts in very early life – according to the Baby Copenhagen substudy, as early as 15 months of age [11]. On the other hand, Lp(a) appears to play a more pronounced role in promoting atheroma development and progression, particularly during the advanced stages of the disease, whereas LDL-C contributes consistently throughout the entire time course of atherogenesis [12, 13].
Unlike LDL-C, Lp(a) levels are not generally modifiable through lifestyle changes (although there is ongoing discussion on Lp(a) level variability), as they are primarily determined by genetic factors [14]. Thus, developing novel therapies is crucial for addressing residual cardiovascular risk, with Lp(a) emerging as a key target for advancing cardiovascular disease prevention. Earlier estimates indicated that lowering Lp(a) by more than 100 mg/dl was necessary to reduce the associated risk of CVD [15]. More recent analyses suggest that a reduction of just 65.7 mg/dl may achieve a comparable decrease in CVD risk to that seen with a 38.7 mg/dl reduction in LDL-C [16].
This review highlights the physiology and pathophysiology of Lp(a), its association with cardiovascular risk, challenges in its measurement, and emerging therapeutics aimed at lowering Lp(a) levels, with a particular focus on the latest findings from 2024. We also explore some of long lingering questions about Lp(a) and its role in cardiovascular disease.
Lipoprotein(A) structure, physiology, and pathophysiology
Lipoprotein(a) is similar to an LDL particle, but it has apolipoprotein(a) [apo(a)] attached to it by both covalent and non-covalent bonding. Apo(a) is a large protein that is primarily synthesized in the liver [17]. A non-covalent bond attaches apo(a) kringle IV domains 7 and 8 to apoB lysine residues; in addition, apo(a) is attached covalently to apoB via a cysteine residue (Cys4057) in the KIV9 domain [18]. Apo(a) is structurally similar to plasminogen, a protein involved in fibrinolysis. Due to this structural similarity, apo(a) may compete with plasminogen when binding fibrin to inhibit fibrinolysis and increase thrombotic risk [18–20]. Plasminogen has 5 kringle domains (KI, KII, KIII, KIV, and KV) and one protease domain at the end, whereas apo(a) has only 10 KIV subtypes (KIV1–KIV10), KV, and a non-reactive serine protease-like domain [19, 20].
Various copies of KIV2 lead to size polymorphism of apo(a), resulting in different levels of Lp(a) in the plasma. Apo(a) isoforms contain between 3 and over 50 KIV2 repeats and have polypeptide molecular masses between ~200 and ~800 kDa [19, 20]. A low number of KIV2 copies (≤ 22) leads to the production of small apo(a) isoforms, which is associated with higher Lp(a) concentrations compared to large apo(a) isoforms (> 22 KIV2 repeats) [21]. When the number of KIV2 repeats is high, a significant portion of apo(a) molecules undergo degradation within hepatocytes before secretion. In contrast, with a low number of KIV2 repeats, the molecules are efficiently secreted and bind to LDL particles outside hepatocytes to form Lp(a) [22]; this may help to understand why Lp(a) is the fifth most prevalent CVD risk factor.
In addition to apo(a) isoform size, which is encoded by the LPA gene, other genetic variants, such as single nucleotide polymorphisms (SNPs), also determine Lp(a) serum levels [23, 24]. Over 2000 SNPs genome-wide are significantly associated with Lp(a) concentrations [23]. Certain SNPs, such as rs1800769 and rs1853021, are associated with lower Lp(a) levels, whereas others, such as rs10455872 and rs3798220, are usually observed with small apo(a) isoforms and are associated with elevated Lp(a) levels [24].
Besides carrying cholesterol esters, free cholesterol, triglycerides, and phospholipids, Lp(a) is the main carrier of oxidized phospholipids (OxPL) among all the apoB-containing lipoproteins [25]. The physiological role of Lp(a) in the body is not fully understood, and its function remains a subject of intense research [26]. However, there are hypotheses regarding its possible roles. Since Lp(a) has no recognized physiological function, it may be associated with a reduced lifespan; this, however, still needs to be confirmed [26].
The high homology between apo(a) and plasminogen (75–99%) suggests a possible role for Lp(a) in fibrinolysis [27]. The latest ex vivo and some human studies do not support the view that Lp(a) is an antifibrinolytic factor [28]. In contrast, in vitro studies have shown that apo(a) potentially affects fibrinolysis in several ways. Apo(a) inhibits (1) plasminogen activation by tissue plasminogen activator (tPA) and (2) the binding of plasminogen to fibrin surfaces. Apo(a) interrupts plasmin-mediated conversion of native Glu1-plasminogen to Lys77-plasminogen. Lys77-plasminogen is a better substrate for tPA, so it is more efficiently converted to plasmin. Inhibition of this conversion by apo(a) leads to a reduction in the amount of plasmin and a weakening of fibrinolysis [27].
The inhibition of fibrinolysis improves wound healing, supporting the theory that Lp(a) may have evolved to have a protective role in primates for certain conditions, such as injuries and infections, particularly during early life [2, 29]. The potential role of Lp(a) in wound healing aligns with immunohistochemical analyses that demonstrated positive staining for apo(a)/apoB in wounds at various stages of healing [30, 31]. Furthermore, a proteomics study identified a correlation between Lp(a) and numerous proteins involved in wound healing [30, 32].
The pathophysiological mechanisms by which Lp(a) contributes to cardiovascular disease risk may involve several interconnected pathways. These include the proatherogenic effects mediated by apoB [33], a pro-inflammatory response driven by OxPL [34], and pro-thrombotic effects resulting from the antifibrinolytic properties of apo(a) [27] and its interactions with platelets [35]. Specifically, processes such as endothelial dysfunction, inflammation, lipid accumulation, and calcification – partly driven by oxidized phospholipids present on the Lp(a) particle – may play a central role in the development of ASCVD and calcific aortic valve stenosis (CAVS) [36, 37].
Lp(a) measurement, nmol/l vs. mg/dl unit
Measurement of Lp(a) is challenging mainly due to the different apo(a) isoforms resulting from the variable number of KIV2 repeats, which can alter its quantification by immunoassays [27]. Additionally, every individual typically inherits and expresses two copies of the LPA gene, one from each parent. Consequently, unless an individual is homozygous for two LPA genes with the same KIV2 repeat count, most people possess two distinct apo(a) isoforms, with their levels representing the combined contributions of both apo(a) isoform sizes [38). Accurate Lp(a) measurement methods are crucial for properly assessing the impact of Lp(a) on various CVDs and for advancing the clinical development of therapies targeting Lp(a). The optimal diagnostic test for Lp(a) should remain unaffected by Lp(a) isoform variability, specifically detect Lp(a) particles, provide results in nanomoles per liter (nmol/l), and rely on standards traceable to globally recognized reference materials [39].
While numerous methods, including immunoassays, fluorescence-based techniques, and electrophoresis, have been developed to measure Lp(a), achieving standardization has proven difficult due to variations in antibody reactivity to different Lp(a) phenotypes [40]. Heydari et al. [40] did not find the assay to be 100% insensitive to apo(a) size. Tests exhibit varying degrees of sensitivity to apo(a) isoforms, which can result in the measured Lp(a) concentration being underestimated in the presence of small isoforms or overestimated when large isoforms are present. However, those assays using 5 to 6 independent calibrators, covering a wide range of Lp(a) levels and a balanced distribution of apo(a) isoforms, are minimally influenced by apo(a) size [36, 41, 42].
The new challenge for Lp(a) measurement arises as a result of the development of muvalaplin – new drug targeting Lp(a) that disrupts the initial non-covalent interaction between apo(a) and apoB100, preventing formation of the disulfide bond and leading to an increase in free apo(a) [43]. Current commercial Lp(a) assays that measure total apo(a) may be insufficient to accurately measure Lp(a) concentrations in patients on this drug; thus, a novel immunoassay that measures only intact Lp(a) particles has been developed [43]. It has been confirmed that this new test is insensitive to apo(a) isoform size and correlates with a liquid chromatography–tandem mass spectrometry method [43].
Measuring Lp(a) in molar units is optimal but poses considerable difficulties. Measuring in mass units (mg/dl) is equally effective, however, for clinical applications [44, 45]. Converting the unit from mg/dl to nmol/l is not recommended, as there is no consistent conversion factor between mass and molar scales due to the varying isoform dependency of each immunoassay-based analytical method [45, 46]. Although the available assays are not yet perfect, most of them can be used for risk stratification of patients [44]. Despite the limitations of mass-based measurement, Lp(a) testing with the most readily available assay (mass- or particle-based) is favored over no testing to facilitate CVD risk stratification, especially in persons with high baseline risk.
This is reinforced by a sub-analysis of the ODYSSEY Outcome trial (Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab), which highlighted that mass and molar lipoprotein(a) immunoassays were similar in their prognostic risk for a major adverse cardiovascular event (MACE) or for MACE reduction with alirocumab. The study included the Siemens N-latex nephelometric immunoassay (measured in mass units – mg/dl), the Roche Tina-Quant turbidimetric immunoassay (measured in molar units – nmol/l), and a non-commercial mass spectrometry (nmol/l) assay [47] (Table I).
Table I
The most burning questions on lipoprotein(a) based on research results published in 2023–2024
Lp(a) concentrations
Plasma Lp(a) levels are approximately 90% genetically determined through the LPA gene [14]. Lipoprotein(a) levels are low at birth, and measurements from umbilical cord blood can be a reliable indicator of neonatal venous blood levels. By 15 months of age, Lp(a) levels reach those typically observed in adults, and the 90th percentile of levels measured at birth (whether from cord or venous blood) serves as a strong predictor for later high-risk levels [11].
Lp(a) concentrations are significantly influenced by race and gender, though the proportional CVD risk due to Lp(a) is roughly similar across ethnicities after considering traditional risk factors [48]. Lower Lp(a) concentrations are found in Chinese, Caucasians, and South Asians, and the highest in Black individuals [2]. The level of Lp(a) is about 17% higher in women than in men after age 50, which is usually associated with the onset of menopause [49]. In women, Lp(a) levels are more variable during their lifetime [50]. Lp(a) levels remain stable from menarche through the reproductive years until the perimenopause. However, during pregnancy, Lp(a) levels approximately double, though the exact mechanisms behind this increase are not understood [51]. It is hypothesized that estrogen may influence Lp(a) synthesis and clearance and that Lp(a) could act as an acute-phase protein in response to endothelial damage, or that it may play a role in placental development [52, 53]. Elevated Lp(a) levels during pregnancy might impact outcomes, potentially raising the risk of complications, such as gestational diabetes, preterm delivery, and low birth weight [54]. Recent data did not confirm the causal relationship between Lp(a) levels and the risk of preeclampsia [55].
Growing evidence has emerged regarding environmental risk factors, conditions, and therapies that can influence Lp(a) levels, potentially contributing to the observed individual variability in Lp(a) [56–58]. Among patients in the placebo group in the OCEAN(a)-DOSE Trial with stable ASCVD and elevated baseline Lp(a) concentrations, notable intraindividual variability of about 10% in Lp(a) levels was observed across all visits [59]. Data from the Nashville Biosciences database showed that baseline and follow-up paired values were significantly different, with an absolute change of ≥ 10 mg/dl in 38.1% and a > 25% change in 40.5% of individuals. Black individuals exhibited greater variability than White individuals; likewise, women exhibited greater variability than men. A positive correlation between the baseline Lp(a) levels and the absolute changes in Lp(a) was also observed; 53% of those in the intermediate “gray zone” category transitioned to either the low-risk (20%) or high-risk (33%) category [60]. In the PMMHRI-Lp(a) Registry, the Lp(a) visit-to-visit variability (mean time distance: 7 ±5 months) was insignificant and only 3.25 mg/dl, but as many as every fourth patient had a difference greater than 10 mg/dl [3]. This issue needs to be further investigated, including the effect of apo(a) isoforms on the level’s variability. This may provide invaluable information for identifying patients who should have Lp(a) measurement more than once [56].
Lp(a) is an acute phase reactant, and is often elevated in conditions such as sepsis, post-surgical states, viral infections, and myocardial infarction [61, 62]. However, the role of Lp(a) as an acute phase reactant remains a subject of ongoing debate [63]. A prospective observational study examined changes in Lp(a) levels in individuals with ST-elevation myocardial infarction (STEMI) at four time points. Median Lp(a) levels increased from 7.9 mg/dl upon hospital admission to 8.4 mg/dl the following day, then to 9.3 mg/dl on the second day (p < 0.001), and further increased to 11.2 mg/dl at 3 months after MI (p < 0.001) [64]. Another study obtained similar results: Lp(a) levels in patients with acute myocardial infarction (AMI) were significantly elevated 6 months after the event, with an increase of at least 25 nmol/l (~10 mg/dl) observed in over 20% of participants. This pattern contrasted with that of high-sensitivity C-reactive protein (hsCRP), suggesting that Lp(a) does not act as a conventional acute phase reactant during AMI [64]. Both studies, however, lack baseline Lp(a) data prior to the occurrence of AMI [64, 65]. This may suggest that repeat testing of Lp(a) after MI should be performed. In total, these findings suggest that repeated measurement of Lp(a) after AMI may be warranted. This concept may be similar to the change in LDL levels after AMI, which in contrast are approximately 10% lower in the peri- and post-MI period [66].
The European Atherosclerosis Society (EAS) consensus statement, the National Lipid Association (NLA), and cardiovascular prevention guidelines from France, Poland, Italy, Canada, India, and China only advise measuring Lp(a) once in a lifetime [45, 56, 67–72]. Polish guidelines do suggest repeating Lp(a) measurements in those patients whose initial Lp(a) levels fall within 30–50 mg/dl (75–125 nmol/l), a range considered the “gray zone” or near the threshold for cardiovascular risk categories. Repeated measurements may be considered for women over the age of 50 years and for patients with chronic kidney disease, particularly nephrotic syndrome, since these conditions can significantly elevate Lp(a) levels [56]. The Polish guidelines [56], as well as the updated NLA recommendations on the use of Lp(a) in clinical practice, recommended selective screening of Lp(a) in high-risk children < 18 years of age in certain cases (based on NLA recommendations in the presence of suspected FH, a first-degree relative with premature ASCVD or elevated Lp(a), or a history of ischemic stroke) [72]. Guidelines also recommend cascade screening of immediate family members of a child or adult with elevated Lp(a) [45, 56, 72]. Recent studies proved that cascade testing for elevated Lp(a) was effective in identifying new cases of elevated Lp(a) [73] (Table I).
Concentrations of Lp(a) and CV risk
CVD risk rises progressively with increasing Lp(a) levels [74, 75], indicating a direct relationship between higher Lp(a) concentrations and elevated cardiovascular risk. However, specific thresholds have been defined and implemented in practice to guide clinical management. Therefore, an Lp(a) level requiring clinical intervention is defined as ≥ 30 mg/dl (≥ 75 nmol/l). Patients with Lp(a) levels between 30 and 50 mg/dl (75–125 nmol/l) fall into a gray zone (also called intermediate risk), which is influenced by both the test’s accuracy and the individual’s overall risk profile [45, 56]. Nonetheless, such individuals already face an elevated risk of adverse events and should be categorized as having moderate risk.
High risk is associated with Lp(a) concentrations above 50 mg/dl (> 125 nmol/l), particularly within the range of 50–180 mg/dl (125–450 nmol/l), while concentrations exceeding 180 mg/dl (> 450 nmol/l) indicate very high risk [45, 56]. There is still an ongoing discussion on the importance of the gray zone in CVD risk prediction. Recent data from the STAR-Lp(a) study showed that the mean CAC-Score in those at low risk (Lp(a) < 30 mg/dl) and patients in the gray zone was almost the same (203.1 ±414.3 vs. 216.6 ±469.4) and essentially increased only for Lp(a) > 50 mg/dl (125 nmol/l) (335.6 ±784.9) [76].
Elevated Lp(a) levels of ≥ 50 mg/dl (≥ 125 nmol/l) are estimated to affect over 1.5 billion individuals globally [2]. Despite this high prevalence, testing for Lp(a) is conducted far less frequently than needed; in most countries, only a few to several percent of patients are tested [77, 78]. A multicenter cross-sectional epidemiological study of 48,135 patients with a history of ASCVD revealed that Lp(a) levels were assessed in only a small proportion of cases (14%) [7]. The prevalence of ASCVD is nearly three times greater in adults with Lp(a) levels above the 99th percentile compared to those with Lp(a) levels at or below the 20th percentile. Among individuals with very high Lp(a), including Lp(a) as a factor leads to the reclassification of one-third of patients in primary prevention and more than half in secondary prevention [79]. Findings from Lp(a) registries highlight the importance of routine Lp(a) measurement, as elevated levels are prevalent among individuals at risk for CVD in primary and secondary prevention settings. This underscores the need for risk re-stratification and treatment optimization in such patients [3–5].
CVD risk and Lp(a)
Elevated Lp(a) levels are associated with increased risk for several cardiovascular diseases including ASCVD [27, 75], aortic stenosis/calcific aortic valve disease (CAVD) [80], ischemic stroke [81], peripheral arterial disease (PAD) [82], heart failure [83, 84], and atrial fibrillation [85]. Based on the data from the Copenhagen General Population Study, Lp(a) levels required to achieve a hazard ratio of 1.5 are 154 nmol/l (~62 mg/dl) for CAVD, 193 nmol/l (~77 mg/dl) for MI, 261 nmol/l (~104 mg/dl) for ischemic stroke, and 323 nmol/l (~129 mg/dl) for HF [72]. Although average or median Lp(a) levels differ globally and across ancestries, the relative risk associated with baseline Lp(a) concentrations appears to be broadly consistent among the populations studied to date [2]. However, the impact of ASCVD attributable to elevated Lp(a) levels may be over twice as high in individuals of African descent compared to those of Caucasian descent [86]. In an analysis of the Women’s Health Study (WHS), the influence of Lp(a), LDL-C, and hsCRP levels on the 30-year risk of CVD events in women was quantified. Elevated Lp(a) levels at baseline were a significant predictor of cardiovascular events over 30 years of follow-up (HR = 1.33, 95% CI: 1.21–1.47). Each of these three biomarkers contributed independently to risk assessment. Considering Lp(a) measurement in addition to traditional markers, such as LDL-C and hsCRP, improves long-term assessment of cardiovascular risk in women [87]. A meta-analysis of randomized controlled trials (RCTs) involving statin therapy including those with and without clinical ASCVD demonstrated that Lp(a) is an independent risk factor across the spectrum of LDL-C, including those with LDL-C < 77 mg/dl [88].
HsCRP is widely recognized as a marker for systemic inflammation and is frequently used in clinical settings to assess inflammatory status and the risk of inflammation-related ASCVD [89]. Previous research showed that elevated Lp(a) was associated with a higher level of hsCRP [3]. Recent studies have investigated the association between Lp(a) levels and the risk of major adverse cardiovascular events (MACE) concerning hsCRP levels. Small et al. found that higher levels of Lp(a) were associated with MACE, MI, and PAD in both primary and secondary prevention populations regardless of baseline hsCRP [90, 91]. Interestingly, Arnold et al. observed that while among individuals without coronary heart disease (CHD) Lp(a) was significantly associated with incident CHD regardless of hsCRP, in participants with CHD at baseline, Lp(a) was related to recurrent CHD events only in those with residual inflammatory risk [92]. Similar results were observed in the analysis from the MESA (Multi-Ethnic Study of Atherosclerosis) study, which identified Lp(a)-associated ASCVD risk only with concomitant elevation of hsCRP [93]. Due to the inconsistency of these studies, this issue merits further investigation [70, 94].
Recently, Alebna et al. [95] conducted a meta-analysis of 562,301 individuals from 11 large cohort studies to clarify this issue. They found that elevated Lp(a) was significantly associated with an increased risk of MACE, regardless of hsCRP levels, in both primary and secondary prevention settings [95]. This discovery highlights that Lp(a) contributes to atherogenesis through various mechanisms and holds significant clinical implications for Lp(a)-targeted therapies, indicating their potential effectiveness in all patients with elevated Lp(a), not just those with concurrently high Lp(a) and elevated hsCRP levels [95].
Coronary artery calcification (CAC) is a hallmark of atherosclerosis and is strongly linked to the overall burden of atherosclerotic plaques [96]. The CAC score is a well-validated metric used to assess atherosclerotic disease burden and guide primary ASCVD management decisions. Findings from the MESA (Multi-Ethnic Study for Atherosclerosis) study revealed that Lp(a) levels and CAC score were independently associated with ASCVD risk and may help guide primary prevention strategies. Participants with elevated Lp(a) and a CAC score > 100 experienced the highest risk (HR = 4.71; 95% CI: 3.01–7.40) compared to those with non-elevated Lp(a) and a CAC score of 0, while individuals with elevated Lp(a) and a CAC score of 0 exhibited a modestly increased risk (HR = 1.31; 95% CI: 0.73–2.35) [97]. The above-mentioned Burzyńska et al. [76] study showed that for each 10 mg/dl (25 nmol/l) increase in Lp(a), the CAC score rose by 15.7 ±0.57 (p = 0.006). In patients with advanced stable coronary artery disease monitored via coronary computed tomography angiography (CCTA) at baseline and after 12 months, elevated Lp(a) levels (> 70 mg/dl) were associated with accelerated progression of the necrotic core, including changes in total, calcific, noncalcific, and low-attenuation plaque [98].
A meta-analysis including 40,073 individuals from 17 studies found that elevated Lp(a) levels were significantly associated with a higher prevalence of CAC (OR = 1.31, 95% CI 1.06–1.61, p = 0.01). When analyzed as a continuous variable, higher Lp(a) levels positively correlated with CAC prevalence (OR = 1.05, 95% CI: 1.02–1.08, p = 0.003). Additionally, elevated Lp(a) was associated with increased progression of CAC over time (OR = 1.54, 95% CI: 1.23–1.92, p = 0.0002) [99].
The relationship between inflammation, Lp(a), and the risk of aortic valve calcification (AVC) was analyzed in a subgroup of patients from the MESA study. The study included 6,676 participants and assessed baseline levels of Lp(a), hsCRP, and AVC using prior non-contrast cardiac computed tomography. Elevated Lp(a) levels were independently associated with AVC, and individuals with both high Lp(a) and elevated hsCRP (> 2 mg/dl) had the highest risk for developing AVC [100]. Lp(a) and its associated molecules – OxPL, autotaxin (ATX), and lysophosphatidic acid (LysoPA) – play critical roles in the development of ASCVD and AVS [101].
Measuring Lp(a) levels in patients with calcific aortic valve disease can help to predict substantially faster disease progression and the likelihood of requiring aortic valve replacement (AVR) [102, 103]. A recent cohort study involving 44,742 patients from a Korean center with Lp(a) level measured from 2000 to 2020, with a mean follow-up of 6.8 years, indicated that AVR due to severe degenerative aortic stenosis was significantly associated with higher levels of Lp(a) (> 100 mg/dl) (adjusted HR = 2.05; 95% CI: 1.31–3.19; p = 0.002) [104]. The authors present compelling evidence reinforcing the association between Lp(a) and advanced CAS, and the necessity for AVR [105].
Aortic valve stenosis, characterized by valvular calcification and stiffness, can lead to heart failure (HF) [106]. Lp(a) has been identified as a possible risk factor for developing heart failure [83, 84]. A meta-analysis of seven Mendelian randomization studies with 300,255 individuals was conducted to explore the causal relationship between Lp(a) and its role in HF. It was demonstrated that increasing Lp(a) levels were significantly associated with increased risk of HF (OR = 1.064, 95% CI: 1.043–1.086, I 2 = 97.59%, p < 0.001) [107]. Moreover, Lp(a) could have a greater impact on HF patients compared to other lipid parameters [89]. The CASABLANCA (Catheter Sampled Blood Archive in Cardiovascular Diseases) study with a total of 1251 individuals indicated that Lp(a) and associated OxPLs may independently contribute to heart injury, leading to HF. Further research, including clinical trials, focused on reducing Lp(a) levels, is needed to determine whether such interventions can prevent the progression to symptomatic HF or mitigate its complications [108].
One of the controversial issues associated with the estimation of CV risk is that cholesterol carried by Lp(a) (Lp(a)-C) is included in both calculated and directly measured LDL-C, as well as in calculated non-HDL-C [109, 110]. Earlier studies suggested that Lp(a) particles contain about 30% cholesterol by mass [109]. However, a recent study involving 68,748 ASCVD-free individuals followed for a median of 9.7 years to track CHD events found that adjusting LDL-C for its Lp(a)-C content did not significantly enhance CHD risk estimation at the population level [111]. The updated NLA guidelines did not endorse the previously proposed correction factor for Lp(a)-C in LDL-C calculations, citing concerns about potentially undertreating high-risk patients [72]. Despite this recommendation, the debate continues. Tsimikas et al. [112] pointed out that the mean Lp(a) level in the Arnold et al. [111] study was only 9.3 mg/dl, and the top decile reached just 43.5 mg/dl – substantially lower than levels associated with elevated ASCVD risk. They emphasized the need for empirically measuring Lp(a)-C and incorporating this adjustment in LDL-C to better understand its impact in observational studies and clinical trials across diverse populations [112] (Table I).
Available lipid-lowering therapies and Lp(a)
Currently, no medications specifically designed to reduce Lp(a) levels have been approved. Consequently, managing elevated Lp(a) involves focusing on reducing overall cardiovascular risk through lifestyle modifications and the intensive management or optimization of other treatable risk factors, by following current clinical guidelines [45, 56, 113, 114].
The findings from studies investigating the impact of statins on Lp(a) levels are inconsistent [25], and the effect seems to be related to apo(a) isoforms. In patients with the low molecular weight apo(a) phenotype, Lp(a) levels increased significantly from 66.4 to 97.4 mg/dl (by 47%; p = 0.026), but not in patients characterized by the high molecular weight apo(a) phenotype [115]. However, even a minor Lp(a) increase of approximately 6–10% following statin use is not clinically significant [116, 117]. Pitavastatin, in contrast to other statins, seems to have a neutral impact on serum Lp(a) levels and may even slightly reduce them; however, this still needs to be confirmed [118]. Ezetimibe and bempedoic acid do not affect Lp(a) levels, as a comprehensive analysis recently found [119–121]. Niacin can lower lipoprotein(a) levels by about 20–30% (depending on the baseline Lp(a) level) due to a decreased LPA mRNA and apo(a) production rate [122]. Again, the amount of reduction seems to be dependent on the size of the apo(a) isoforms [56]. However, no clinical benefit was noted with this therapy, so its use is no longer currently recommended [123, 124]. The prior CVD outcome trials involving niacin were also not enriched for individuals with elevated Lp(a).
Proprotein convertase subtilisin/kexin type 9 inhibitors (PCSK9is) and small interfering RNAs (inclisiran) were found to decrease circulating Lp(a) by ~30%, as demonstrated in various studies including the FOURIER, ODYSSEY Outcomes, and ORION-11 trials [125–129]. Post-hoc analyses of the FOURIER and ODYSSEY Outcomes trials demonstrated that the 3-year number needed to treat (NNT) to prevent one recurrent ASCVD event with PCSK9 monoclonal antibodies is at least 2.5-fold lower for individuals with higher versus lower levels of Lp(a) [125, 126]. These drugs, however, are not approved for this purpose, making it difficult to obtain reimbursement for their use in lowering Lp(a). Significant variability in response to these drugs is also observed, with participants who had higher baseline plasma Lp(a) levels experiencing greater absolute reductions in Lp(a) [127–130]. The size of apo(a) seems to be an independent determinant of the response to PCSK9is, with each additional kringle domain being associated with a 3% additional reduction in Lp(a) (the larger the isoforms, the lower the Lp(a) level, the better the response) [130].
A significant number of patients on PCSK9 inhibitors are concurrently treated with statins and/or ezetimibe. However, the combined impact of these therapies on Lp(a) levels, as well as the relationship between apo(a) isoform size and the Lp(a) response, remains unclear. A recent prospective study evaluated lipid lowering in participants with an LDL-C > 100 mg/dl who received evolocumab 140 mg combined with either atorvastatin 80 mg or ezetimibe 10 mg daily. The findings revealed variability in Lp(a) reduction, with changes in Lp(a) levels being strongly associated with apo(a) isoform size [131].
Lipoprotein apheresis is currently the only method capable of significantly lowering Lp(a) levels [132, 133]. A single apheresis session can reduce Lp(a) concentrations by approximately 60–75%, while regular treatments every 1–2 weeks result in a sustained reduction of around 25–40% from baseline levels [134, 135]. Lipoprotein apheresis in patients with ASCVD and elevated Lp(a) leads to a notable decrease in cardiovascular events [136]. The US Food and Drug Administration (FDA), as well as European guidelines [56], approved apheresis for patients with Lp(a) levels exceeding 60 mg/dl (> 150 nmol/l), regardless of baseline LDL-C levels [137]. However, this treatment approach is invasive, expensive, time-consuming for the patient, has limited availability, and is often associated with a diminished quality of life [138] (Table I).
Aspirin and Lp(a)
Aspirin is one of the most well-established therapies for secondary prevention of ASCVD related events, and based on recent data it may also be beneficial in primary prevention for patients with elevated plasma Lp(a). Large RCTs of aspirin use for primary prevention, including ARRIVE (Aspirin to Reduce Risk of Initial Vascular Events), ASCEND (A Study of Cardiovascular Events in Diabetes), and ASPREE (Aspirin in Reducing Events in the Elderly), conducted in different populations, found no significant net beneficial effect of aspirin on CVD events [139–141]. A meta-analysis of 13 RCTs with 164,225 participants and 1,050,511 participant-years of follow-up indicated a small benefit of aspirin use for CVD risk in primary prevention [142].
Post-hoc analyses of the ASPREE trial have shown that individuals with elevated Lp(a) genotypes may derive a net benefit with aspirin therapy [143]. In a recent propensity-matched cohort study (MESA), aspirin use was associated with a significant reduction (46%) in risk for cardiovascular events among individuals with Lp(a) > 50 mg/dl and without baseline cardiovascular disease [144]. The patients with Lp(a) > 50 mg/dl and aspirin use had similar CHD risk as those with Lp(a) ≤ 50 mg/dl, regardless of aspirin use [144]. This was the first study to focus on Lp(a) level measurements, contrary to earlier studies that used the SNP of LPA. These results align with earlier evidence showing a 45–55% reduction in the risk of initial MACE among regular aspirin users who carry the rs3789220 LPA gene variant, while no such benefit was observed in non-carriers [143, 145].
Moreover, the results from the MESA study were further supported by data from the National Health and Nutrition Examination Survey (NHANES III) analysis, which gathered baseline data from 1988 to 1994 in a nationally representative cohort of US adults without clinical ASCVD. Their findings revealed that regular aspirin use was independently associated with a 52% reduction in ASCVD mortality risk among individuals with elevated Lp(a), but this association was not observed in those without elevated Lp(a) during a median follow-up period of 26 years [146]. Interestingly, in the post-hoc analysis of the PEGASUS-TIMI 54 trial, individuals with elevated Lp(a) ≥ 200 nmol/l (80 mg/dl) and a history of MI (within 1 to 3 years) may possibly more strongly benefit from dual antiplatelet therapy (DAPT) with ticagrelor in the setting of background aspirin [147].
While bleeding events were not evaluated in that study, the MESA study reported a higher bleeding rate among aspirin users (17.5% vs. 12.5%, p < 0.01). However, after multivariable adjustments, no correlation was observed between bleeding rates and Lp(a) levels. Consequently, additional research is necessary to assess the overall (net) clinical benefit in this context [148].
There is a lack of effective therapies specifically aimed at reducing cardiovascular disease risk in individuals with elevated lipoprotein(a), particularly for primary prevention. In the just-published recommendations of Polish Experts endorsed by the Polish Lipid Association (PoLA), aspirin is not recommended for primary prevention in patients with low or moderate cardiovascular risk, regardless of the co-occurrence of elevated Lp(a) concentration. However, the use of aspirin in primary prevention should be considered (class IIa recommendation) in patients with at least high cardiovascular risk with elevated Lp(a) levels > 50 mg/dl (> 125 nmol/l), which may help optimize the risk of ASCVD associated with it [149] (Table I).
New therapies for lowering Lp(a)
Although there is a pressing demand for therapies specifically designed to lower Lp(a) levels, the sole available targeted treatment option at present is apheresis. No FDA-approved medications exist for this purpose yet, but several innovative treatments to lower Lp(a) are under investigation in late-stage randomized controlled trials [150].
Several RNA-based therapies against Lp(a) are under development. The success of the first agent – pelacarsen, an antisense oligonucleotide (ASO) – paved the way for the development of further small interfering RNAs (siRNAs) agents, such as olpasiran, zerlasiran, and lepodisiran [41, 151–153] (Table II). The mechanism of action of ASOs and siRNAs is to reduce Lp(a) levels by inhibiting apo(a) protein synthesis [154]. Maximum reported Lp(a) reduction for this drug was 80% for pelacarsen and 98% for olpasiran, zerlasiran, and lepodisiran [150].
Table II
Lp(a)-lowering drugs under clinical development
[i] ASCVD – atherosclerotic cardiovascular disease, ASO – antisense oligonucleotides, CAD – coronary artery disease; CETP – cholesterol ester transport proteins, CV – cardiovascular, FH – familial hypercholesterolemia, PAD – peripheral artery disease, SC – subcutaneous, siRNA – small interfering RNAs, T2DM – type 2 diabetes mellitus.
Pelacarsen, given as an 80 mg subcutaneous injection once a month, is currently undergoing a phase 3 clinical trial to evaluate its impact on reducing CVD event risk (Lp(a) HORIZON trial, (NCT04023552). The trial has completed enrollment of over 8,000 patients but is expected to conclude in 2026 because of the low event rate [155]. Moreover, pelacarsen is also under investigation in the CAVS trial, a phase 2 randomized controlled study (NCT05646381) designed to evaluate its ability to slow the progression of aortic stenosis (AS) compared to placebo. The trial targets patients with mild to moderate AS and elevated Lp(a) levels, plans to recruit around 500 participants, and is projected to conclude in 2029 [156].
Recently reported results of the phase 2 OCEAN(a)-DOSE off-treatment extension period indicated that olpasiran demonstrates long-lasting effects in reducing Lp(a) levels, with participants who received doses of ≥ 75 mg every 12 weeks maintaining approximately a 40% to 50% decrease in Lp(a) levels nearly 1 year after their final dose [151]. Olpasiran, developed by Amgen, is currently being evaluated in the phase 3 clinical trial known as the Ocean(a) study (NCT05581303), which has enrolled approximately 7,000 participants. The findings, anticipated in 2027, are expected to follow the results of the HORIZON study by about a year and will shed light on olpasiran’s impact on cardiovascular events [157]. Also worth noting is a forthcoming RCT with olpasiran in primary prevention: A Double-blind, Randomized, Placebo-controlled, Multicenter Study Assessing Olpasiran Use to Prevent First Major Cardiovascular Events in High-risk Participants with Elevated Lipoprotein(a). It is of critical importance, as most patients with elevated Lp(a) are currently diagnosed in primary CVD prevention, still before the event.
Lepodisiran, developed by Eli Lilly, has completed its phase 1 trial. Its phase 2 trial (the Alpaca Phase 2 trial; NCT05565742) is currently underway and is expected to end in October 2024 [152, 158]. The results of this lepodisiran trial with extended duration up to 540 days are to be presented at the ACC Congress in Chicago in March 2025. Undergoing a phase 3 clinical trial, ACCLAIM Lp(a) (NCT06292013), is projected to be the largest study of Lp(a)-lowering therapies, aiming to enroll 12,500 participants, with an expected completion date in 2029 [159]. The ACCLAIM trial is another study that will allow evaluation of the effect of Lp(a) reduction on MACE both in adults with elevated Lp(a) who have established ASCVD and in those in primary prevention who are at risk for a first CVD event [159].
The last siRNA drug, zerlasiran, has completed its phase 1 clinical trial and a phase 2 trial (NCT05537571). The results were released at the American Heart Association Scientific Sessions in Chicago 2024 and published simultaneously. Zerlasiran achieved over an 80% reduction in time-averaged lipoprotein(a) levels over 36 weeks when administered at 300 mg every 16 weeks or 300 mg and 450 mg every 24 weeks. Sustained reductions in Lp(a) levels were observed up to 60 weeks after the initial dose, and no safety concerns were identified. These results support advancing zerlasiran to a phase 3 trial in its development program [152]. Lepodisiran and zerlasiran have similar effects as olpasiran, with an over 90% Lp(a) reduction at the highest doses, with comparable tolerability and safety profiles [152, 153].
In 2024, there was a major breakthrough in developing a new strategy for lowering Lp(a) levels. Results of phase II clinical research with the first oral agent, muvalaplin, were published, and the new therapy based on gene editing was revealed in preclinical studies [160, 161]. Muvalaplin, developed by Elly Lily, is a small molecule inhibitor of Lp(a) synthesis, lowering Lp(a) levels by binding to apo(a) KIV7 and KIV8. This interaction sterically hinders the covalent attachment of apo(a) to apoB, thereby reducing Lp(a) formation [162]. Findings from a phase I study (NCT04472676) demonstrated that the treatment was well tolerated and achieved a maximum reduction in Lp(a) levels ranging from 63% to 65% [160]. The phase II KRAKEN trial with muvalaplin (NCT05563246) enrolled 233 participants (median age 66 years) with lipoprotein(a) ≥ 175 nmol/l and ASCVD, diabetes, or familial hypercholesterolemia. Treatment was for 12 weeks. Muvalaplin was well tolerated and caused placebo-adjusted reductions in lipoprotein(a) of 47.6% (95% CI: 35.1–57.7%), 81.7% (95% CI: 78.1–84.6%), and 85.8% (95% CI: 83.1–88.0%) for the dose of 10 mg/day, 60 mg/day, and 240 mg/day, respectively [163].
Available commercial Lp(a) assays measure total apo(a), apo(a) in the Lp(a) particle, and apo(a) that is not bound to apoB, and may be insufficient to accurately measure Lp(a) concentrations, especially after muvalaplin treatment, taking into consideration its mechanism of action where apo(a)–muvalaplin complexes in circulation might be detected. Swearingen et al. [43] introduced an innovative immunoassay designed to measure only Lp(a) particles. This particle-specific electrochemiluminescent (ECL) immunoassay employs an anti-apo(a) capture antibody that targets a common epitope found in KIV7, KIV8, and KIV9, ensuring isoform insensitivity. Detection is achieved using an anti-apoB monoclonal antibody, preventing the assay from identifying unbound apo(a). A recent study evaluated the Lp(a)-lowering effects of two therapeutics with distinct mechanisms of action: lepodisiran and muvalaplin. The results revealed that the commercial assay measuring total apo(a) underestimated the Lp(a)-lowering efficacy of muvalaplin compared to the intact Lp(a) assay, which specifically measures Lp(a) particles. However, the Lp(a)-lowering impact of lepodisiran was found to be clinically comparable between the intact Lp(a) assay and the commercial assay [43].
CTX320 is an experimental CRISPR/Cas9-based gene editing therapy designed to target and disable the apo(a) component of Lp(a) production in the liver. The therapy utilizes lipid nanoparticles to deliver Cas9 mRNA and guide RNA (gRNA) directly into the body. CTX320 has the potential to permanently reduce Lp(a) after a one-time treatment [161]. Preclinical studies in non-human primates demonstrated that CTX320 reduced Lp(a) levels in a dose-dependent manner, achieving approximately 20%, 80%, and 90% reductions from baseline at doses of 0.5, 1.5, and 3 mg/kg, respectively [155]. In a separate ongoing study, a single infusion of CTX320 at 2 mg/kg resulted in a ~94% reduction in Lp(a) levels by day 14, with this reduction maintained until day 224. The therapy was well tolerated in these studies and showed a durable lowering of plasma Lp(a). As a result, CTX320 is being prepared for advancement to phase I clinical trials [164].
Obicetrapib, a next-generation selective inhibitor of cholesteryl ester transporter protein (CETP), is currently undergoing clinical trials to lower LDL-C levels and reduce major adverse cardiovascular events. The results of the phase 2 ROSE trial (NCT04753606) demonstrated that obicetrapib, administered orally at doses of 5 mg and 10 mg alongside intensive statin therapy, reduced Lp(a) levels by 33.8% and 56.5%, respectively, compared to placebo [165]. The phase 2 ROSE1 and ROSE2 trials investigated obicetrapib as an add-on therapy to high-intensity statins in individuals without CVD but with LDL cholesterol levels > 70 mg/dl. A pooled analysis revealed that obicetrapib 10 mg, combined with high-intensity statin therapy, significantly reduced Lp(a) levels by 57% compared to placebo. This reduction surpasses those achieved with PCSK9 inhibitors (15–30%), niacin (30%), or other CETP inhibitors (25%) [166]. At the AHA 2024, the results of the BROOKLYN trial with obicetrapib in patients with heFH (baseline Lp(a) level in the obicetrapib group was 45.8 nmol/l) were released, showing a 54.3% placebo-adjusted Lp(a) reduction in the intervention arm and 38% of patients with > 50% Lp(a) reduction [167]. Currently, obicetrapib is under investigation in a phase 2 trial (VINCENT). It is an open-label, 16-week trial aimed at evaluating Lp(a) levels for patients with elevated Lp(a) being treated with obicetrapib and obicetrapib/evolocumab. The VINCENT trial started recruiting patients at the end of 2024 and will be completed at the end of 2025 (NCT06496243) [168] (Table II).
Safety of very low levels of Lp(a)
Observational and epidemiological studies have suggested that extremely low Lp(a) levels, typically below 7 mg/dl, are associated with an elevated risk of type 2 diabetes mellitus (T2DM), as has been shown for LDL lowering with statin treatment [169–171]. However, the underlying mechanism and causality of this relationship have not been established [172, 173], leaving open the question of whether novel Lp(a)-lowering therapies might contribute to an increased risk of developing T2DM [174].
The results of a Mendelian randomization (MR) analysis published in 2024 shed more light on this association. Data from a two-sample Mendelian randomization analysis involving data from 563,420 patients from the UK Biobank and FinnGen consortia did not show a correlation between Lp(a) and T2DM [175]. Another study with a two-sample MR analysis of the UK Biobank population cohort also found no evidence for an association between genetically predicted Lp(a) and T2D [176]. Moreover, two-sample MR analysis using summary-level genome wide association data suggested that hyperinsulinemia, often associated with type 2 diabetes mellitus, may partially explain the inverse association observed between low lipoprotein(a) levels and an increased risk of developing type 2 diabetes mellitus [177]. High fasting insulin levels, which contribute to the progression of prediabetes and type 2 diabetes, are actually responsible for the observed reduction in Lp(a) level. Therefore, while a connection between Lp(a) and diabetes does exist, it is unlikely that Lp(a) serves as a risk factor for diabetes (no reverse causality) independent of the presence of underlying hyperinsulinemia and insulin resistance [177]. Thus, aggressive Lp(a)-lowering therapy at this time does not raise any substantial concerns about exacerbated T2DM risk (Table I).
Conclusions and take-home message
In 2024, knowledge about the importance of Lp(a) as a risk factor for cardiovascular diseases was deepened, and great progress was made in developing therapies to effectively lower its levels, which may contribute to reducing the risk of these diseases in the future [178, 179]. While most clinical trials with new Lp(a)-lowering therapies focus on ASCVD patients or those at CVD risk, only one started in 2024 will evaluate the ability of pelacarsen to slow the progression of aortic stenosis. Moreover, there was a breakthrough in therapy for lowering Lp(a) levels based on gene editing. Currently, Lp(a) is utilized in clinical practice to refine risk stratification and guide patients toward more aggressive risk factor management. In the future, it may be possible to implement targeted treatments specifically aimed at lowering Lp(a) levels. However, the physiological functions of Lp(a) are still largely unknown. Although some safety concerns over lowering Lp(a) to very low levels remain, recent Mendelian randomization studies did not find an association between very low levels of Lp(a) and new onset T2D. Lipoprotein(a) research is a rapidly evolving field, but many questions remain unanswered. We eagerly await the results of the HORIZON, OCEAN and ACCLAIM studies to establish whether or not Lp(a) lowering will transform our approach to the prevention of ASCVD (Figure 1).