Introduction
Without effective vaccination, progressive hepatitis C virus (HCV) represents a major global health problem. It predisposes to the development of hepatic and extrahepatic complications and mortality [1, 2]. Worldwide, particularly among the population aged 15 years or over, Egypt has the highest prevalence. Therein, the prevailing genotype is 4, which is the most aggressive and highly associated with severe fibrosis and transformation [3]. Despite the accumulation of a large body of data, HCV pathogenesis requires further scrutiny.
One pathogenic mechanism for HCV is oxidative stress resultant from increased generation of reactive oxygen species in infected hepatocytes, where redox homeostatic balance is a major challenge to prevent cell death and DNA mutation [4–8]. Mechanistically, HCV activates prooxidant enzymes and hepatic stellate cells, weakens antioxidant defenses, and induces organelle damage, secretion of pro-inflammatory cytokines and metals unbalance – secondary to mitochondrial failure, which is a focal point in that scenario [9, 10]. The early induced oxidative stress is responsible for HCV liver disease progression towards both steatosis and fibrosis [9].
Liver injury disturbs hepatic architecture and hinders blood flow that is further deteriorated by the locally induced vaso-/angioactive compounds. The resultant multiple areas of hypoxia increase lactate accumulation and reduce its clearance. In such a scenario, hypoxia seems to be a systemic as well as a local problem, as reflected by the induced systemic angiogenesis [11–16]. Hypoxia-induced vascular endothelial growth factors (VEGF) and their counteracting soluble receptors (sVEGFRs) are major pathophysiological players. Their inducibility is orchestrated by the transcription regulator, the hypoxia-inducible factor-1α (HIF-1α) [17–20]. HCV infections promote oxidative stress, while downregulating VEGF mRNA expression very early after infection that rebounds in the long term [9]. The marked variability in VEGF and sVEGFR concentrations in HCV-induced hepatic disease makes them weak indicators for the progression of chronic liver disease in cirrhotic patients. However, they may reflect the increases in portal hypertension and/or the decreases in hepatic regenerative activity [18–21]. Moreover, the interaction between the virus and the immune system response is a critical pathogenic mechanism. In chronic liver disease, abnormal immunological responses include autoantibodies, hypergammaglobulinemia, cytokine profile changes and oxidative stress, especially in decompensated hepatic cirrhosis [21].
Diagnostic blood markers and non-invasive imaging methods for determining liver damage and its pathogenesis are being validated. The target of this study was to investigate the changes in oxidative stress, cellular hypoxia/angiogenesis and immunological biomarkers in blood of Upper Egyptian patients with treatment-naïve HCV infection (with normal vs. high enzyme levels) and liver cirrhosis compared to healthy volunteers. The correlations between these biomarkers and the liver state were analyzed as potential pathogenic effectors and/or diagnostic biomarkers.
Material and methods
Setting
This cross-sectional study voluntarily enrolled consented patients and their age- and socio-economically matching healthy companions attending the Hepatology Institute, Assiut University Hospitals, Assiut, Egypt, in the period from January 1, 2016 to December 1, 2018. The local Bioethics Committee of the Faculty of Medicine, Assiut University approved this study. De-identified demographic data and disease history were collected from patients’ records. Patients’ exclusion criteria included immobilization for any reason, renal failure, autoimmune diseases, acute infections, congenital and hemolytic anemia and systemic inflammatory diseases.
370 treatment-naïve patients with HCV-related chronic liver disease were consecutively included in our study. They encompassed three groups: 120 with normal serum liver enzymes (HCV-NE; mean ± SDM, ALT = 32.75 ±10.91; AST = 35.75 ±7.01; APL = 70.29 ±15.19 U/l), 130 with high serum enzymes (HCV-HE; ALT = 120.5 ±83.97; AST = 85.1 ±45.87; APL = 120.5 ±44.85 U/l; p < 0.001), and 120 cirrhotic patients. Hospital-based diagnosis – through thorough clinical examination, abdominal ultrasonography, liver function tests and liver biopsy – was done to characterize the chronic liver disease. HCV infection was diagnosed based on specific positive anti-HCV serological testing and RT-PCR assessed positive HCV RNA. Forty healthy controls were enrolled for comparison. The male-female ratio in all groups was 1 : 1.
Samples and investigations
Serum was recovered from fasting peripheral blood samples collected in plain tubes after clotting and centrifugation. Serum was aliquoted and stored at –70°C. Plasma recovered from another fasting heparin/sodium fluoride peripheral blood sample was immediately deproteinized with two volumes of perchloric acid for enzymatic measurement of lactate (Cat. No. 16300, Greiner Diagnostic GmbH, Bahlingen, Germany). Serum total IgG was measured using a SPAPLUS kit (Code NK004.S, The Binding Site Group Ltd, Birmingham, UK). Using a specific commercially available ELISA kit, serum granulocyte-macrophage colony-stimulating factor (GM-CSF) was measured (Cat. No. KAC0901, BioSource Europe S. A., Nivelles, Belgium). Quantikine VEGF immunoassay kits were used to measure serum VEGF and sVEGFR1 (Cat. # RRV00 and # DVR100B, R&D Systems, Inc., MN, USA). Serum total peroxides were measured colorimetrically as H2O2 equivalents by xylenol orange reagent [22]. We used 2,2′-azinobis-3-ethylbenzothiazoline-6-sulfonate (ABTS) for the colorimetric measurement of serum total antioxidants (TAOs) as Trolox equivalents [23]. Serum oxidative stress index (OSI) was calculated as a percent ratio of TAO content in mM to total peroxides in mM of each sample [22]. Serum malondialdehyde (MDA), a lipid peroxidation and oxidative stress biomarker, was assayed colorimetrically using 2-thiobarbituric acid as 1,1,3,3-tetraethoxypropane precursor equivalents [24].
Statistical analysis
Data are presented as frequency (n) and mean ± SDM. Prism 6.0 GraphPad (GraphPad Software, Inc., La Jolla, CA, USA) was used for the statistical analysis. One-way ANOVA with the Newman-Keuls multiple comparison test was applied. Correlation among parameters within groups was analyzed using Spearman’s nonparametric correlation analysis. The significance limit was set at a p-value of ≤ 0.05 at a 95% confidence interval.
Results
Changes in oxidative stress biomarkers
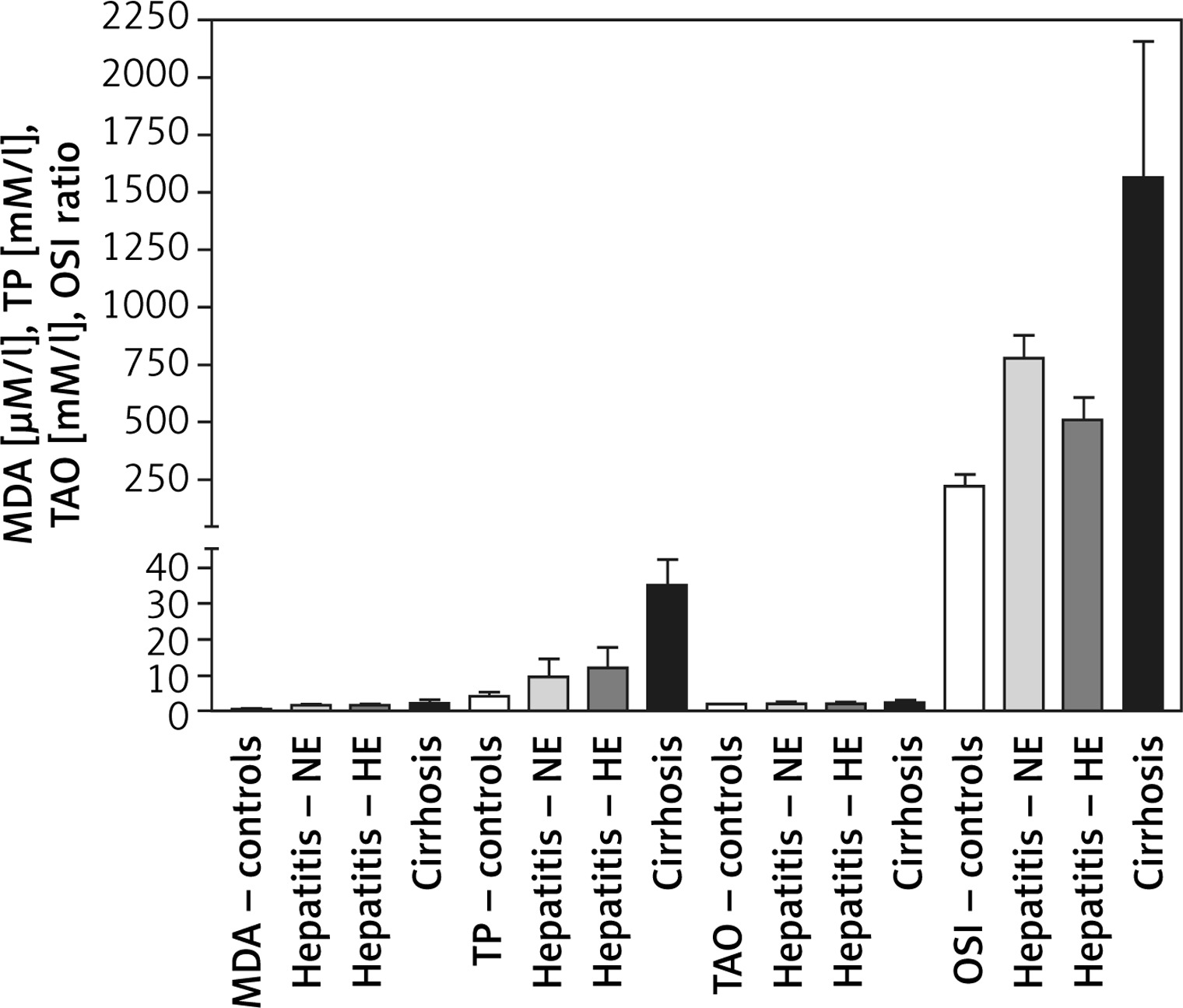
MDA levels were 0.56 ±0.12, 1.6 ±0.21, 1.62 ±0.37, and 1.934 ±0.622 μM/l for the healthy controls, HCV-NE, HCV-HE, and cirrhotic patients, respectively. This revealed markedly higher MDA levels in the three patient groups compared to the healthy controls (p < 0.001). There were no significant differences among the patient groups. Total peroxide levels for controls, HCV-NE, HCV-HE and cirrhotic patients were 4.0 ±1.0, 9.41 ±5.08, 12.11 ±5.64, and 35.1 ±7.303 mM/l, respectively. This showed significantly higher total peroxides in all patient groups vs. healthy controls (p < 0.001). A small, significant increase was observed comparing HCV-NE and HCV-HE patients (p < 0.05). Cirrhotic patients revealed markedly higher levels of total peroxides vs. the other patient groups (p < 0.001). TAOs levels of 1.89 ±0.1, 2.03 ±0.54, 1.89 ±0.56, and 2.249 ±0.611 mM/l for the healthy controls, HCV-NE, HCV-HE, and cirrhotic patients (respectively) were non-significantly different. OSI was 220 ±52.6, 778.4 ±100.8, 508.6 ±98.16, and 1560.7 ±595.3 for controls, HCV-NE, HCV-HE, and cirrhotic patients, respectively. Because of the marked increase in total peroxides, this OSI was significantly lower in controls vs. each of HCV-NE, HCV-HE, and cirrhotic patients (p < 0.001, 0.05, and 0.001, respectively). HCV-NE and HCV-HE groups were significantly different (p < 0.05), and each of them was significantly lower than cirrhotic patients (p < 0.001) (Figure 1).
Figure 1
Changes in serum oxidative stress biomarkers of Egyptian patients with HCV infection (with normal vs. high enzyme levels) and liver cirrhosis vs. healthy controls. Data shown are mean ± SDM
MDA – malondialdehyde, TP – total peroxides, TAO – total antioxidants, OSI – oxidative stress index, NE – HCV infection with normal serum enzymes, HE – HCV infection with high serum enzymes.
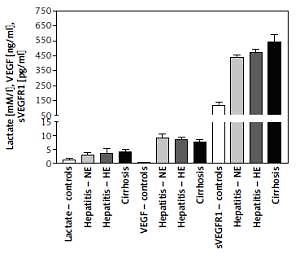
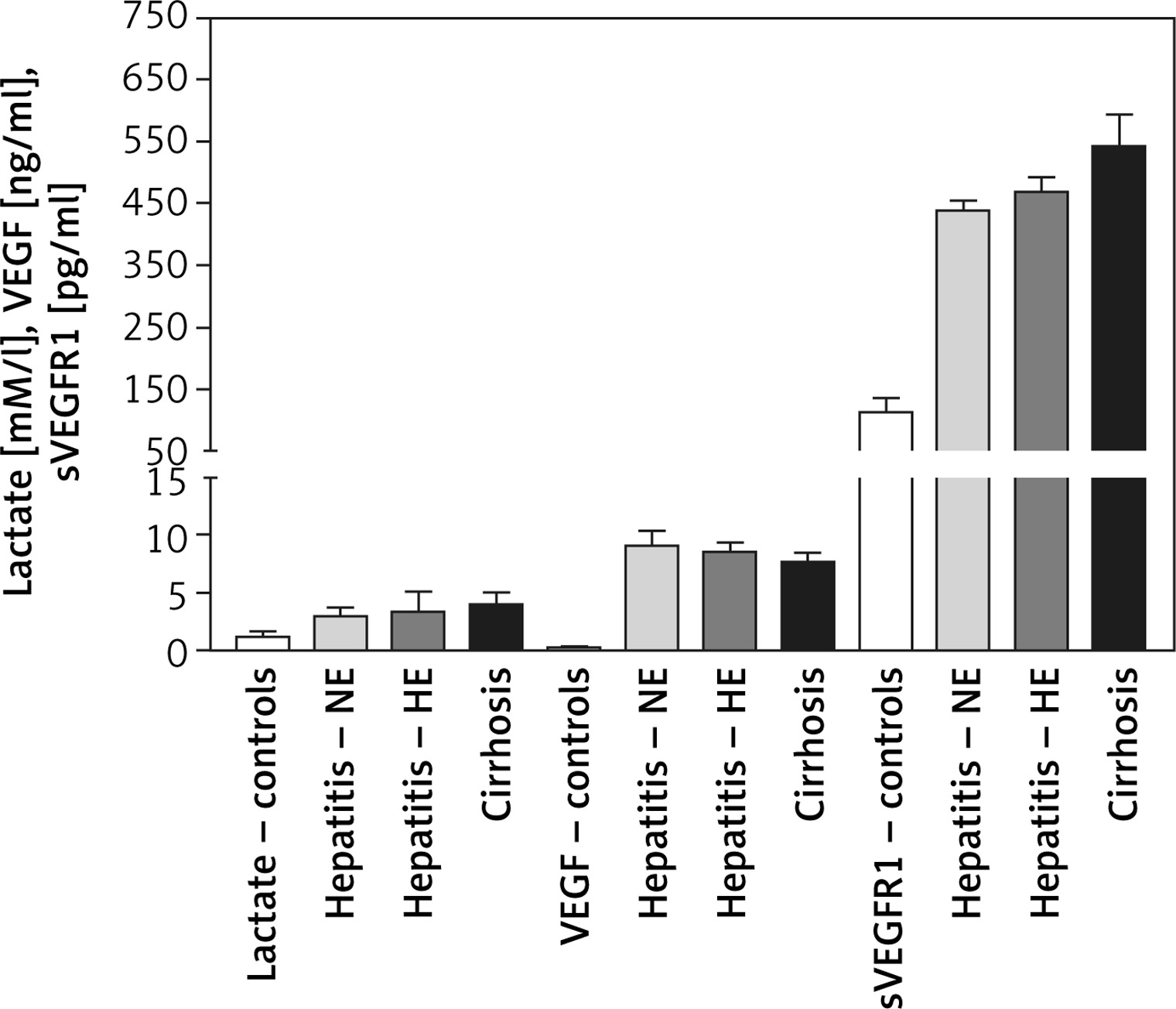
Changes in blood hypoxia and angiogenesis biomarkers
Plasma lactate levels were 1.3 ±0.447, 2.98 ±0.74, 3.39 ±1.71, and 3.959 ±1.127 mM/l for controls, HCV-NE, HCV-HE, and cirrhotic patients, respectively. Lactate was significantly lower in controls compared to all patient groups (p < 0.001). Patient groups were non-significantly different. VEGF levels were 0.22 ±0.08, 9.1 ±1.27, 8.56 ±0.79, and 7.705 ±0.785 ng/ml in controls, HCV-NE, HCV-HE and cirrhotic patients, respectively. This indicated the massively increased levels in all patient groups compared to controls (p < 0.001). Compared to HCV-NE and HCV-HE groups, cirrhotic patients had lower VEGF levels (p < 0.001 and 0.05, respectively). In controls, HCV-NE, HCV-HE and cirrhotic patients the serum sVEGFR1 levels were 114 ±22, 439.1 ±15.35, 469 ±23.9, and 542 ±52 pg/ml, respectively. This demonstrated that sVEGFR1 levels were more than tripled in all patient groups vs. controls (p < 0.001). Significantly higher levels of serum sVEGFR1 were noted in the cirrhotic patients than the other two patient groups (p < 0.001). HCV-HE patients exhibited higher serum sVEGFR1 levels compared to the HCV-NE group (p < 0.05) (Figure 2).
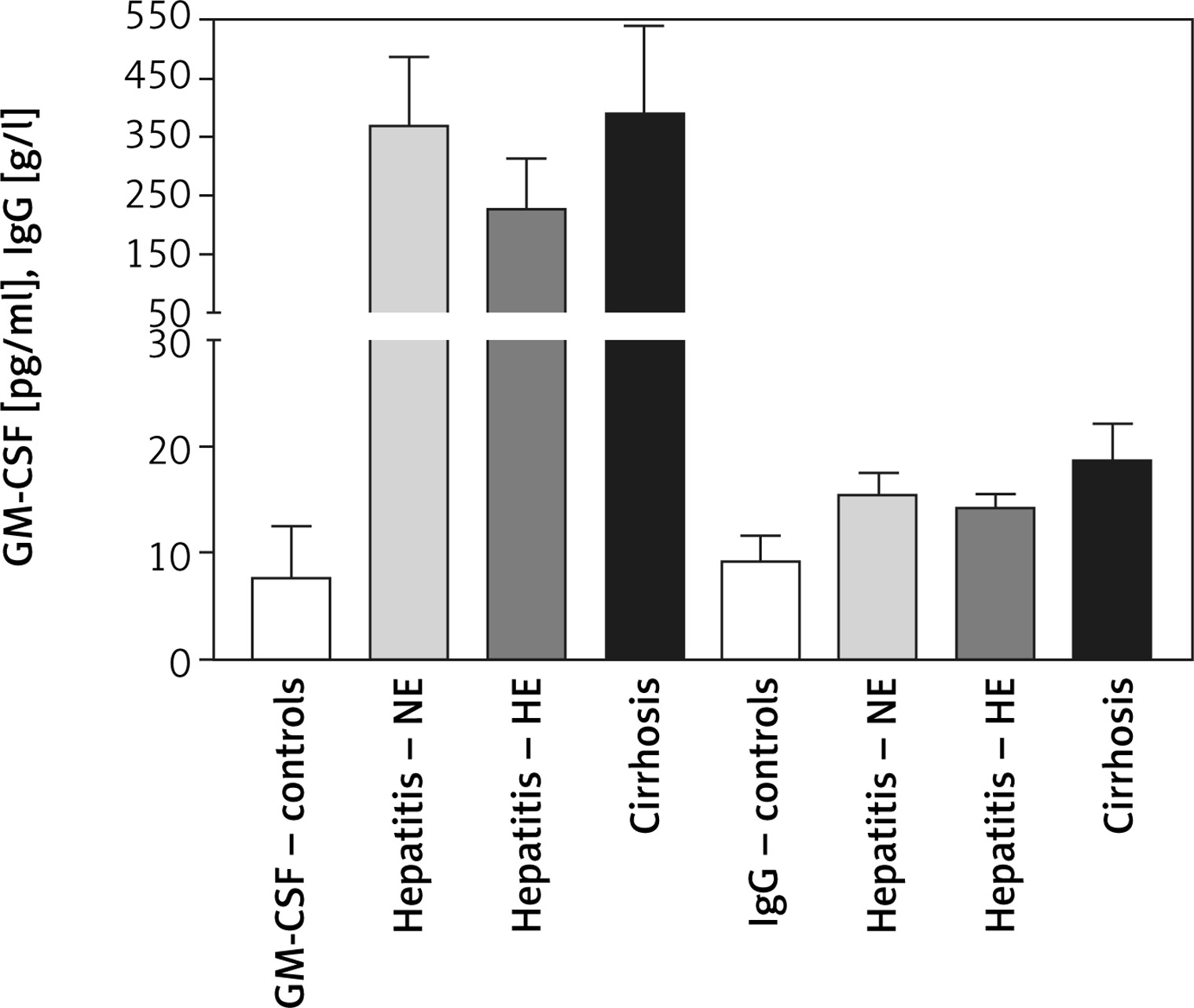
Changes in serum immunological biomarkers
Serum GM-CSF levels were 7.62 ±4.92, 370 ±117, 227 ±87, and 388 ±152 mM/l for controls, HCV-NE, HCV-HE, and cirrhotic patients, respectively. This indicated that serum GM-CSF level was increased to more than 25-fold the control levels in the patient groups (p < 0.001). HCV-NE and cirrhotic patients had a higher GM-CSF level vs. HCV-HE patients (p < 0.01). Serum total IgG levels in controls, HCV-NE, HCV-HE and cirrhotic patients were 9.299 ±2.358, 15.4 ±2.1, 14.2 ±1.37, and 18.58 ±3.596 g/l, respectively. Patient groups showed markedly higher levels of IgG than controls (p < 0.001). Compared to both HCV groups, cirrhotic patients had significantly higher levels of serum total IgG (p < 0.001), and patients with HCV-HE showed significantly lower contents compared to patients with HCV-NE (p < 0.01) (Figure 3).
Correlation analysis among parameters
Correlations among all parameters were nonsignificant in healthy controls. In HCV-NE patients, total peroxides and MDA correlated positively (r = 0.643, p < 0.05), and sVEGFR1 vs. IgG (r = –0.689, p < 0.05) and TAOs vs. MDA (r = –0.664, p < 0.05) showed significant negative correlations. In HCV-HE patients, there were significant positive correlations between lactate and each of VEGF (r = 0.622, p < 0.05), GM-CSF (r = 0.718, p < 0.01) and IgG (r = 0.689, p < 0.05), between VEGF and GM-CSF (r = 0.660, p < 0.05), between sVEGFR1 and each of GM-CSF (r = 0.602, p < 0.05) and IgG (r = 0.522, p < 0.05), and between TAOs and IgG (r = 0.484, p < 0.05). In cirrhotic patients, there were significant positive correlations between VEGF and each of sVEGFR1 (r = 0.650, p < 0.05), total peroxides (r = 0.619, p < 0.05) and lactate (r = 0.700, p < 0.05); between TAOs and each of total peroxides (r = 0.887, p < 0.01) and GM-CSF (r = 0.676, p < 0.05), and between lactate and each of sVEGFR1 (r = 0.733, p < 0.05) and MDA (r = 0.778, p < 0.01).
Discussion
The blood-borne hepatitis C virus infection is the most common cause of chronic viral hepatitis in the world, including Egypt [25]. In the absence of an in vitro system that recapitulates the in vivo stages of HCV infection, the pathogenic mechanisms leading to the hepatocellular damage are not fully understood. However, it is established that not only does HCV infection induce oxidative stress, but also the replication of the HCV virus depends on such redox-sensitive cellular changes [26]. Biomarkers of oxidative stress are increased in liver tissue and body fluids of patients with chronic HCV infection. Induction of oxidative stress correlates with the incidence and/or severity of HCV-related morbidities, since chronic HCV infection relies on this strategy for immune escape and persistence [5–8]. In support, antioxidants were promising co-adjuvants in HCV therapy [27]. In the same direction, our results revealed marked oxidative stress in HCV patients caused mainly by increases in oxidants rather than decreases in antioxidants. MDA level was significantly higher in patient groups compared to controls, which in part reflects the continuing cellular damage in response to HCV. Reportedly, MDA level and its protein adducts are elevated in liver and blood samples of chronic HCV patients and correlated positively with the stage of hepatic fibrosis and negatively with the response to treatment [28]. Moreover, ischemia-reperfusion, which simulates hypoxia and the HIF-1α-orchestrated response, increases tissue MDA and induces pronounced changes in hepatic architecture due to steatosis and cell destruction [15]. Similarly, our patients also showed a significant increase in OSI due to the increased total peroxides, and HCV-NE patients revealed a significant positive correlation between MDA and total peroxides. This was reasoned to be due to inflammation and mitochondrial dysfunction [29]. Two possible explanations for the non-significant changes in TAOs among all groups are: 1) induction of antioxidant mechanisms because of the disease to counteract the generated oxidative stressors, and 2) the increases in the highly antioxidant bilirubin in patients. TAOs and MDA showed a significant negative correlation in HCV-NE patients similar to previous studies [6, 9, 11, 28]. In the cirrhotic patients, there was a positive significant correlation between TAOs and total peroxides. Therefore, the redox homeostatic imbalance caused the pro-oxidant status and oxidative stress in our patients despite the increases in antioxidants. Reportedly, reactivation of HCV viral replication could favor the prooxidant state that exacerbates cell damage and death [26].
Lactate is produced in the peripheral tissues under normal physiological conditions, and is metabolized mainly in the liver and kidneys. HCV infection induces transcriptional downregulations that affects mainly mitochondrial respiratory chain complex core subunit genes. This induces metabolic reprogramming and aerobic glycolysis with lactate accumulation and enhanced generation of reactive oxygen species [11, 16]. HCV viral-protein expression and infection severely impair mitochondrial oxidative phosphorylation leading to major reliance on nonoxidative glucose metabolism. This occurs through induction of HIF-1α and its dependent VEGF and glycolytic enzymes, in vitro and in vivo [30–32]. Interestingly, such hypoxic conditions and associated gene expression changes enhance HCV replication. Vice versa, inhibition of HIF-1α reduced HCV replication; therefore, they are pathogenic in the disease process [12–14]. Consequently, prolongation of lactate elimination in cirrhotic patients compared to normal individuals is rationally expected [33]. Moreover, such simple lactemia turns into aggressive lactic acidosis upon HCV antiviral treatment [34]. In agreement, our observation of a significant increase in blood lactate and a positive correlation between lactate and MDA in cirrhotic patients indicate that they both reflect hepatocellular damage [33].
Experimental models of portal hypertension and liver cirrhosis inferred a direct role for angiogenesis. Among a long list of angiogenic modulators, VEGF is the most potent angiogenic and vasculogenic mediator. The cellular signaling machinery is exploited by human viruses to facilitate their own pathogenesis through upregulating the expression of VEGF [35]. VEGF is up-regulated in experimental and clinical cirrhotic livers. HCV infection accelerates angiogenesis by stabilizing HIF-1α under normoxic growth conditions that correlates with increased VEGF secretion and membrane receptor expression [36–38]. Our study demonstrated that, compared to healthy controls, our HCV patients show significantly higher serum levels of VEGF but serum aminotransferases did not correlate with VEGF in patients. Consequently, it was established that the enhanced regenerative capacity of hepatocytes rather than the hepatocyte damage per se is the cause of the increased serum VEGF [30, 31]. The clinical utility of sVEGFR1 as a pathogenic, diagnostic and/or prognostic marker in different medical conditions, including liver cirrhosis, either separately or accompanied with VEGF, was previously reported [39]. Our results are in concordance with such a notion; we observed elevated VEGF and over 3-fold increase in its antagonistic sVEGFR1 in all patient groups vs. controls. These changes are associated with the degree of hepatic insufficiency [39]. We found significant positive correlations in the cirrhotic patients between VEGF and each of total peroxides and lactate. This indicates that the triad of oxidative stress, hepatocellular dysfunction and angiogenesis/hypoxia are intertwiningly implicated. Similarly, we found significant correlations between lactate and VEGF in HCV hepatitis with high serum enzymes. Moreover, we observed significant positive correlations in the cirrhotic group between VEGF and sVEGFR1, and between lactate and sVEGFR1. These results confirmed the earlier findings of a dramatic elevation in VEGFA serum levels in HCV patients. Similar findings connected angiogenesis, as overexpressed VEGFA transcripts, with the progression of chronic liver diseases into fibrosis [40–42]. In cirrhotic patients as compared to healthy controls, over 3-fold increases in VEGF and sVEGFR1 levels were reported. Patients with the advanced stages of liver cirrhosis recorded the highest concentrations [39]. Serum sVEGFR1 increased to more than 3-fold the healthy control level in HCV-infected patients with hepatocellular carcinoma and correlated with the liver enzyme level and tumor stage in Egyptian patients [17]. Egyptian HCV-infected patients showed dramatically elevated serum levels of VEGF compared to healthy controls [43]. Compared to healthy controls, HCV patients had significantly higher VEGFA in peripheral blood mononuclear cells and serum [19]. Such levels confirmed the other international studies and correlated with the fibrosis grade in HCV-induced liver fibrosis [19, 20]. Egyptian patients with HCV-related liver cirrhosis had about 3-fold increases in serum VEGF that positively correlated with liver enzyme levels [44]. Chronic HCV with cirrhosis revealed significantly lower VEGF but higher sVEGFR1 levels compared to controls [18]. There was no significant difference comparing serum VEGF levels in patients with chronic HCV infection and those with HCV-related liver cirrhosis [45]. In patients with chronic hepatitis and liver cirrhosis, serum sVEGFR-2 correlated with the disease severity [46]. Egyptians with genotype-4 chronic HCV infection showed a moderate but significant increase in serum VEGF [19]. A Turkish study reported more than 3-fold increase in serum VEGF in HCV infection and liver cirrhosis patients that inversely correlated with fibrosis and cirrhosis scores [21]. The discrepancies among these reports could be related to variations in the disease course (early vs. chronic), viral load and activity, virus genotype and genetic background of patients.
HCV-induced progressive liver disease deteriorates from chronic inflammation to fibrosis and cirrhosis. Such a persistent uncontrolled inflammatory state worsens the hepatic damage. Changes in the inflammatory chemokine and cytokine profiles parallel the progress of HCV infection and its response to treatment. Therefore, changes in chemokines and cytokines are being validated as biomarkers for disease progression, its therapeutic responses and drug targets [47]. Moreover, the natural course of infection and the outcome of antiviral treatment are determined by the nature of the cytokine response against HCV [48]. Because of that, coupling of the direct-acting anti-HCV drugs with anti-inflammatory/hepatoprotective agents yields a better therapeutic prognosis [47].
GM-CSF regulates inflammation, resistance to viral infections and stimulation of tissue repair [49–51]. It stimulates the production of granulocytes and monocyte-macrophages [52]. GM-CSF serum level was increased to more than 25-fold the healthy control levels in our patients. These results are in line with a previously reported Egyptian study that showed significant elevation in GM-CSF among chronic HCV patients [53]. Moreover, the increases in GM-CSF were correlated with increased lactate accumulation [54]. Interestingly, our results showed significant correlations between GM-CSF and each of lactate, VEGF, and sVEGFR1 in HCV patients with high serum enzymes. Furthermore, our cirrhotic patients revealed a significant positive correlation between GM-CSF and TAOs. Reportedly, compared to subjects in whom therapy was unsuccessful, the pretreatment cytokine response (including GM-CSF) was higher in patients who achieved a sustained virological response. Levels of histological grading and serum enzymes also positively correlated with the expression of GM-CSF, among other cytokines [48]. While serum enzymes were associated with the histologic activity index, GM-CSF was associated with a change in fibrosis stage [55]. However, a Brazilian study showed lower serum GM-CSF levels in chronic HCV patients than healthy controls. This may reflect the prevailing genotype and natural course of the disease [56]. GM-CSF has pro-inflammatory (granulocyte and macrophage growth and differentiation) and anti-inflammatory/regulatory (dendritic cell differentiation and increased regulatory T-cell numbers) effects. However, these effects depend on the presence and dose of other relevant cytokines in the immune response milieu. GM-CSF enhances the anti-HCV immune response induced by the HCV nonstructural protein NS3 [57]. This implicates the changes in the immunological cytokine profile as the fourth dimension in the pathogenic vicious tetrad of highly hepatotoxic feed-forward cycle of inflammation and cell death – alongside oxidative stress, hepatocellular dysfunction and angiogenesis/hypoxia. Reportedly, the abnormal immunological responses, as a feature of chronic HCV-induced liver disease, include autoantibodies and hypergammaglobulinemia – especially in decompensated hepatic cirrhosis [58]. Hypergammaglobulinemia is pathogenically causative in the progression of liver disease and advanced forms of hepatic fibrosis [59, 60]. In support, total serum levels of IgG in our study showed significantly higher levels in all patient groups compared to controls. Along with the futile increased production, this could be due to reduction in the rate of clearance of immunoglobulins by the diseased liver [61]. Interestingly, in HCV-HE hepatitis, there were significant positive correlations between IgG and each of lactate, TAOs, and sVEGFR1, indicating the intimate interaction of the four pathogenic mechanisms investigated in our study. In the milder HCV-NE hepatitis, the correlation analysis revealed negative significant correlations between sVEGFR1 and IgG.
In conclusion, we have demonstrated the intimate pathogenic interactions among the significantly induced oxidative stress, immunological, and cellular hypoxia/disordered angiogenesis in our HCV hepatitis and cirrhosis patients. Our data further highlighted the potential prognostic and druggable value of most of these biomarkers, particularly sVEGFR1 in HCV-induced liver fibrosis.