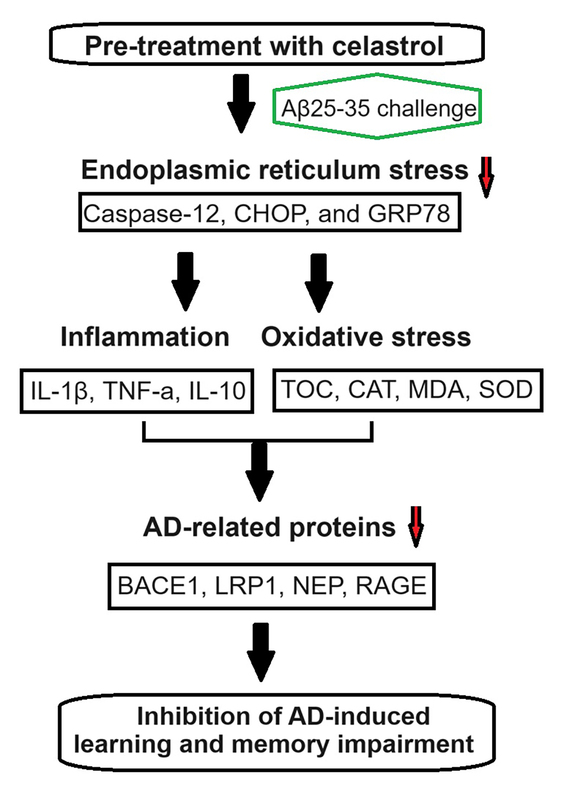
Introduction
Alzheimer’s disease (AD) is characterized by progressive degeneration of the nervous system [1]. It involves the buildup of neurofibrillary tangles and neuritic plaques resulting from amyloid-β (Aβ) peptide accumulation in brain regions such as the neocortex and medial temporal lobe [2–4]. Clinical manifestations of AD include cognitive decline, behavior alterations, emotional volatility, and sleep disruptions, culminating in severe symptoms such as malnutrition, multi-organ failure, and even brain death [5, 6]. Despite these challenges, a specific cure for AD remains elusive. Various genetic, epigenetic, and environmental factors contribute to its development.
Various mechanisms, including AD, trigger neuroinflammation and oxidative stress. Inflammation can compromise the blood-brain barrier, intensifying oxidative stress and neuronal damage [7, 8]. Elevated levels of interleukins are associated with cognitive impairment, alongside increased oxidative stress marked by reactive oxygen species (ROS) production, reduced antioxidants and elevated malondialdehyde [9–12]. Oxidative stress accelerates aging, damages neurons, and increases susceptibility to conditions such as AD [13]. Numerous agents have been used to control neuroinflammation; however, their efficacy remains limited. Celastrol, derived from Tripterygium wilfordii, exhibits anti-inflammatory effects by inhibiting NF-κB activity and regulating cytokine release [14–17], with reports on its potential protective role in AD models.
The endoplasmic reticulum (ER) is crucial for cellular functions such as protein synthesis, calcium storage, and lipid metabolism [18]. The ER maintains intracellular calcium balance; ER disruption triggers ER stress and molecular chaperone expression to restore cellular homeostasis [19]. Researchers have described the contributory role of ER stress in AD progression [20]. For instance, Huangpu Tongqiao Capsule exerts neuroprotective effects against AD by targeting ER stress-induced apoptosis [21], whereas luteolin mitigates cognitive deficits in AD mouse models by inhibiting ER stress-dependent neuroinflammation [22]. Therefore, ER stress regulation appears promising for AD treatment. In this study, we elucidated the influence of celastrol treatment on learning and memory decline in the AD mouse model. Our results show that celastrol may attenuate learning and memory impairment in AD animal models.
Material and methods
Chemical and reagents
The following primary antibodies were utilized: anti-BACE1 antibody (Abcam, ab183612); anti-LRP1 antibody (Abcam, ab92544); anti-MCK10/NEP antibody (Abcam, ab216341); anti-RAGE antibody (Abcam, ab216329); anti-Caspase-12 antibody (Abcam, ab315271); anti-GRP78 BiP antibody (Abcam, ab21685); anti-GAPDH antibody (Abcam, ab9485). The following substances were employed: tunicamycin (Sigma, T7765); sodium tauroursodeoxycholate (TUDCA) (Sigma, T0266). Celastrol was sourced from Aktin Chemical (Chengdu, China) with a purity exceeding 98%.
Preparing the animal model
C57BL/6 mice (half male, half female) aged 8 weeks were obtained from the Shanghai Model Organisms Center. Half of the mice were randomly assigned to five groups as follows: control, AD, and three celastrol-treated AD groups (pre-injection, 1 mg/kg, 3 mg/kg, and 6 mg/kg). The other half of the mice were also randomly assigned to five groups: control, AD, and three celastrol-treated AD groups (post-injection, 1 mg/kg, 3 mg/kg, and 6 mg/kg). Each group consisted of 12 mice, all born on the same day, with 6 males and 6 females. The environmental conditions were consistent across all groups, and the experimenters were blinded to the treatments.
AD was induced in the mice based on previously established protocols with minor adjustments [23]. The mice were anesthetized with phenobarbital and positioned in a stereotaxic apparatus (RWD Life Science, China). A midline sagittal incision was created, and coordinates for the hippocampal CA1 region were determined using a standard brain stereotaxic atlas as follows: anteroposterior –3.5 mm from bregma, mediolateral ±2.0 mm from the midline, and dorsoventral –2.8 mm from the skull [24]. After marking these points on the skull, 2 μl of sterile normal saline solution consisting of 10 μg of aggregated amyloid beta-peptide 25-35 (Aβ25-35) (5 μg/μl) (Sigma, USA) was injected bilaterally into the CA1 region of the dorsal hippocampus. All animal experiments were conducted following the NIH guideline Guide for the Care and Use of Laboratory Animals. The animal study protocol was approved by the Laboratory Animal Ethic Committee of Gongli Hospital of Shanghai Pudong New Area (No. LAEC2022-0316).
Drug administration
In the pre-injection celastrol-treated AD groups, the mice were administered celastrol intraperitoneally (1, 3, 6 mg/kg/day) for 2 days beginning 1 day before Aβ25-35 microinjection. In the post-injection celastrol-treated AD groups, the mice were administered celastrol intraperitoneally (1, 3, 6 mg/kg/day) for 2 days beginning 14 days after Aβ25-35 microinjection. The dosages were chosen based on previous studies [25]. In some experiments, the mice were administered tunicamycin (100 nM) or tauroursodeoxycholic acid (TUDCA) (1 μM) intraperitoneally in a similar manner.
Barnes maze cognitive test
After fourteen days of exposure to experimental conditions, the mice underwent the Barnes maze cognitive test to evaluate their spatial learning and memory [26]. The maze consists of a circular platform with 20 evenly spaced holes, one of which leads to a dark chamber called the goal box. The test involved placing the mice in the center of the maze and encouraging them to locate and enter the goal box using aversive noise and bright light. The spatial learning training lasted for 5 days, with each trial lasting 3 min. Four trials were conducted daily, with a 15-minute break in between each one. Memory tests were conducted on days 7, 14, and 21 to assess short-term, medium-term, and long-term retention. The time taken for the mice to enter the target box was recorded for each trial using the ANY-Maze video tracking system (SD Instruments, USA).
Morris water maze test
The Morris water maze test was conducted according to established procedures using a circular tank that was 150 cm in diameter and 50 cm in height, had a water depth of 30 cm, and maintained a temperature of 20°C to 24°C [27]. Chinese ink was used to hide the platform’s location, which was a plastic platform submerged 1.5 cm below the water’s surface in each of the four equal quadrants of the maze [28]. Visual cues were placed on the walls to assist with spatial orientation, and testing took place between 9 a.m. and 12 p.m. under controlled conditions.
Each group of mice participated in four trials per day, with each trial lasting a maximum of 120 s, separated by 10-minute intervals over a period of 5 days starting on day 4 after receiving the celastrol microinjection. The trials were followed by a 20-minute break for 5 consecutive days. The test trial took place 1 h after the completion of the fifth training day. Detailed analysis was conducted on parameters including latency to find the platform, path length, time spent in each quadrant, and platform crossings.
Western blot analysis
Protein was extracted from mouse brain tissues using the radioimmunoprecipitation assay buffer (Beyotime, China) to determine the protein quantity and concentration with a bicinchoninic acid assay (Beyotime, China). The samples were then loaded onto a 12% gel (Bio-Rad, USA), subjected to electrophoresis, transferred onto a 0.45 μm PVDF membrane (GE Healthcare, USA), and blocked with nonfat dry milk (Beyotime, China) after suitable dilution. Primary antibodies were left to incubate overnight at 4°C, followed by secondary antibodies for 2 h at 25°C. The imaging was done using the ChemiDoc MP Imaging System (Bio-Rad, USA) with a chemiluminescent HRP substrate (Millipore, USA). The antibodies used are shown in Table I.
Table I
Antibodies table
Enzyme-linked immunosorbent assay
Mice were deeply anesthetized and decapitated after the behavioral test in order to collect brain tissue promptly. The tissue was ground thoroughly into a homogenate using phosphate-buffered saline (PBS) and centrifuged at 10,000 rpm for 15 min. The resulting supernatant was carefully collected and used for conducting the competitive enzyme-linked immunosorbent assay (ELISA) according to established protocols [29]. ELISA kits for IL-1β, TNF-α, and IL-10 (Elabscience, China) were used in accordance with the manufacturer’s instructions.
Measuring total antioxidant capacity, catalase, malondialdehyde, and superoxide dismutase levels
After the behavioral test, mice were deeply anesthetized and decapitated for the oxidative stress analysis. The brain tissue was quickly collected, homogenized with PBS, and centrifuged at 12,000 rpm for 30 mins. The resulting supernatant was used for further analysis. The Mouse TAC ELISA Kit (Abcam, UK) and Mouse Cathepsin ELISA Kit (Abcam, UK) were used to determine the levels of total antioxidant capacity (TAC) and catalase (CAT), respectively. The MDA assay kit (Abcam, UK) was utilized to measure the malondialdehyde (MDA) level. The Mouse Superoxide Dismutase ELISA Kit (Abcam, UK) was employed to quantify the superoxide dismutase (SOD) level.
HE staining, Nissl staining, and immunohistochemistry
Before staining, all slides were prepared through a series of steps. Initially, they were exposed to 3% H2O2 for 10 min. Following that, the slides were subjected to microwaving in 0.01 M citrate buffer (pH 6.0) for 10 min to retrieve antigens. Subsequently, they were blocked with 10% normal goat serum to prevent any non-specific binding. Hematoxylin and eosin staining involved staining the slides with the Hematoxylin and Eosin Staining Kit (Solarbio, China) for 1 h at room temperature. For Nissl staining, the slides were stained using the Nissl Staining Kit (Solarbio, China) for 20 min at 25 °C.
During immunohistochemistry, the slides were left to incubate overnight at 4°C with primary antibodies, followed by incubation with mouse/rabbit HRP-conjugated secondary antibodies (Dako, Denmark) for 1 h at room temperature. Subsequently, they underwent 3,3′-diaminobenzidine substrate color development for immunostaining detection. Lastly, the tissues were counterstained with hematoxylin to improve contrast. Image capture was done using an Axio Scope A1 optical microscope (Zeiss, Germany), and areas positive for immunostaining were quantified and analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, USA). The results were expressed as relative integrated optical density, calculated as the sum of staining intensity divided by the total area.
Statistical analysis
Statistical analysis was performed using SPSS 17.0 software (SPSS Inc, USA). Quantitative data were analyzed using a one-way analysis of variance, followed by multiple comparison Dunnett post hoc tests to determine statistical significance. Results are presented as mean ± standard deviation. The comparison was considered significant when p < 0.05.
Results
Celastrol attenuated AD-induced learning and memory impairment
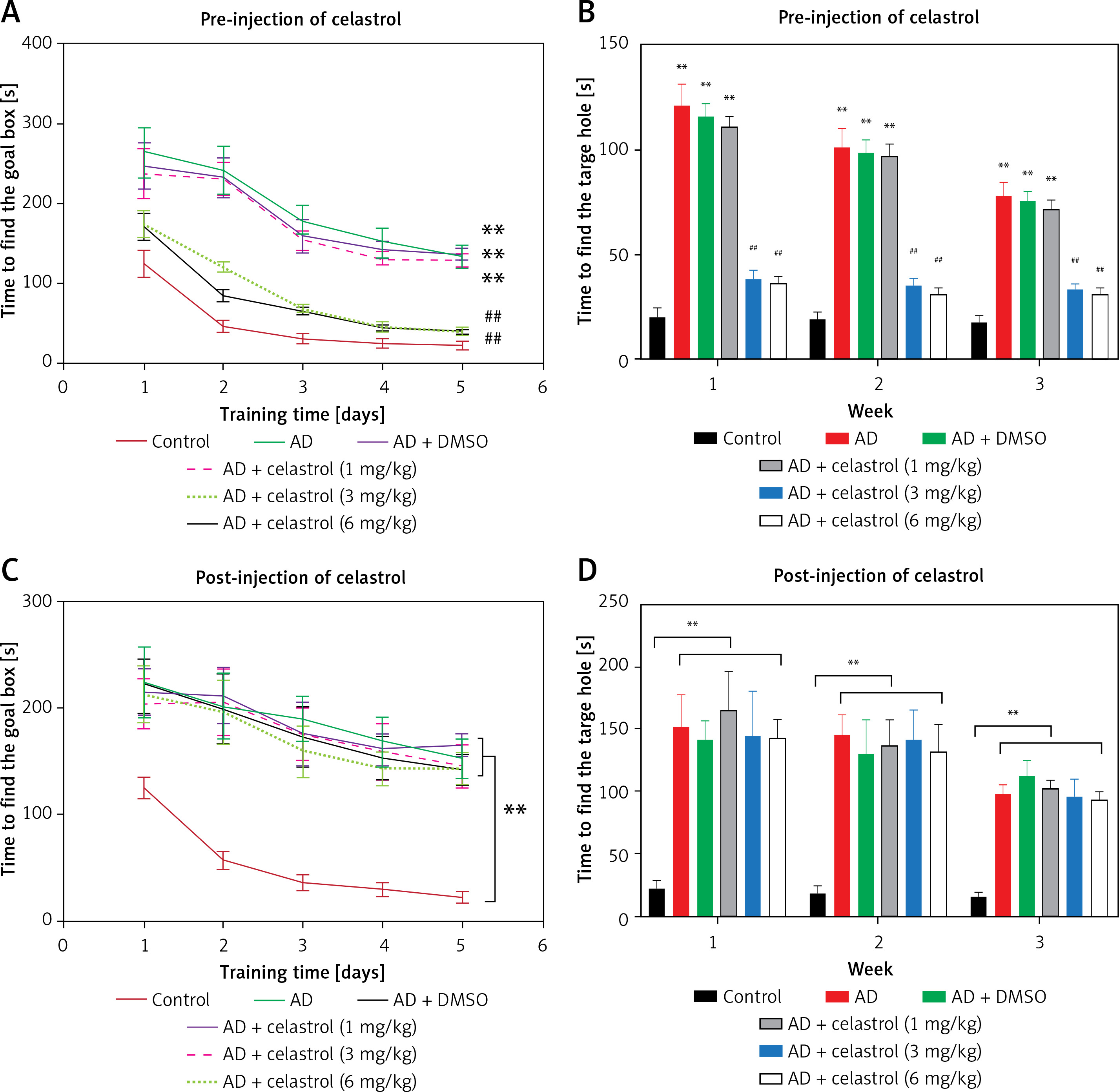
The Barnes maze cognitive test was carried out on 12 mice in each group – control, AD, and celastrol pretreated AD (1 mg/kg, 3 mg/kg, and 6 mg/kg) – to assess physical deficits following AD induction in mouse models. The test was performed daily after a training period, with significantly shorter times recorded on days 2, 3, and 4 compared to day 1 for all groups (Figure 1 A). AD mice took longer to learn and reach the goal box compared to controls, but treatment with celastrol, especially at 3 mg/kg, mitigated the effects of AD. Therefore, AD mice required more time to locate the goal box during training. Celastrol treatment reversed the learning and memory deficits induced by AD, and also reduced the time taken to find the goal box after training sessions (1, 2, and 3 weeks) in AD model mice (Figure 1 B). However, according to Figures 1 C and D, post-injection of celastrol did not significantly reverse the AD-induced impairments observed during training to locate the goal box. The time to find the goal boxes and target holes was not notably affected by post-treatment with celastrol (1 mg/kg, 3 mg/kg, and 6 mg/kg), indicating that post-treatment with celastrol was unable to reverse the AD-induced learning and memory impairments.
Figure 1
Celastrol pre-treatment attenuates Alzheimer’s disease (AD)-induced learning and memory impairment (Barnes maze cognitive test) in mice. A – Training performance during the Barnes maze cognitive test with celastrol pre-treatment. B – Memory phase performance during the Barnes maze cognitive test with celastrol pre-treatment. C – Training performance during the Barnes maze cognitive test with celastrol post-treatment. D – Memory phase performance during the Barnes maze cognitive test with celastrol post-treatment. Error bars represent the means ± SEM of the three independent experiments; **P < 0.01 compared to Control; ##P < 0.01 compared to AD + DMSO
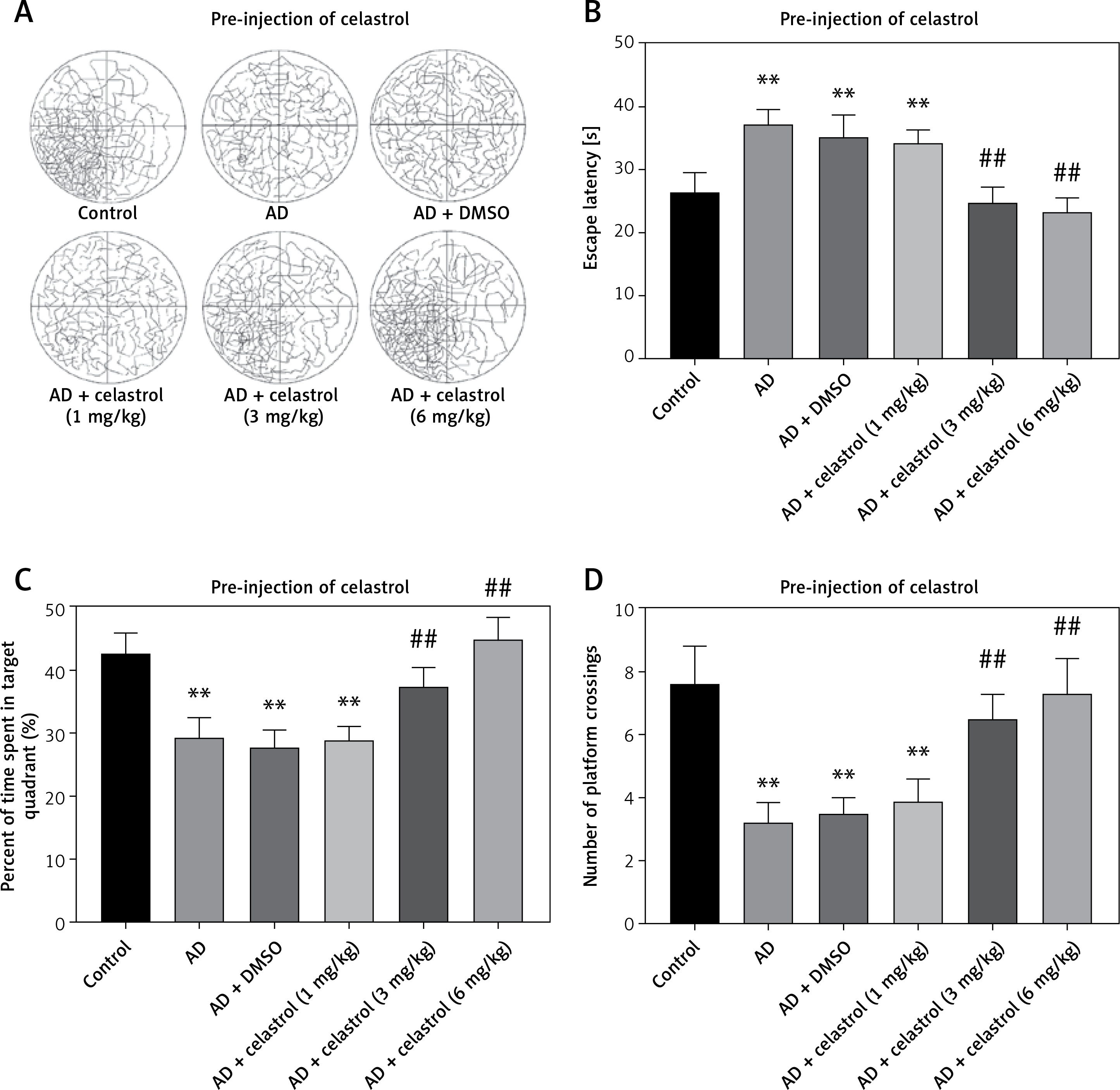
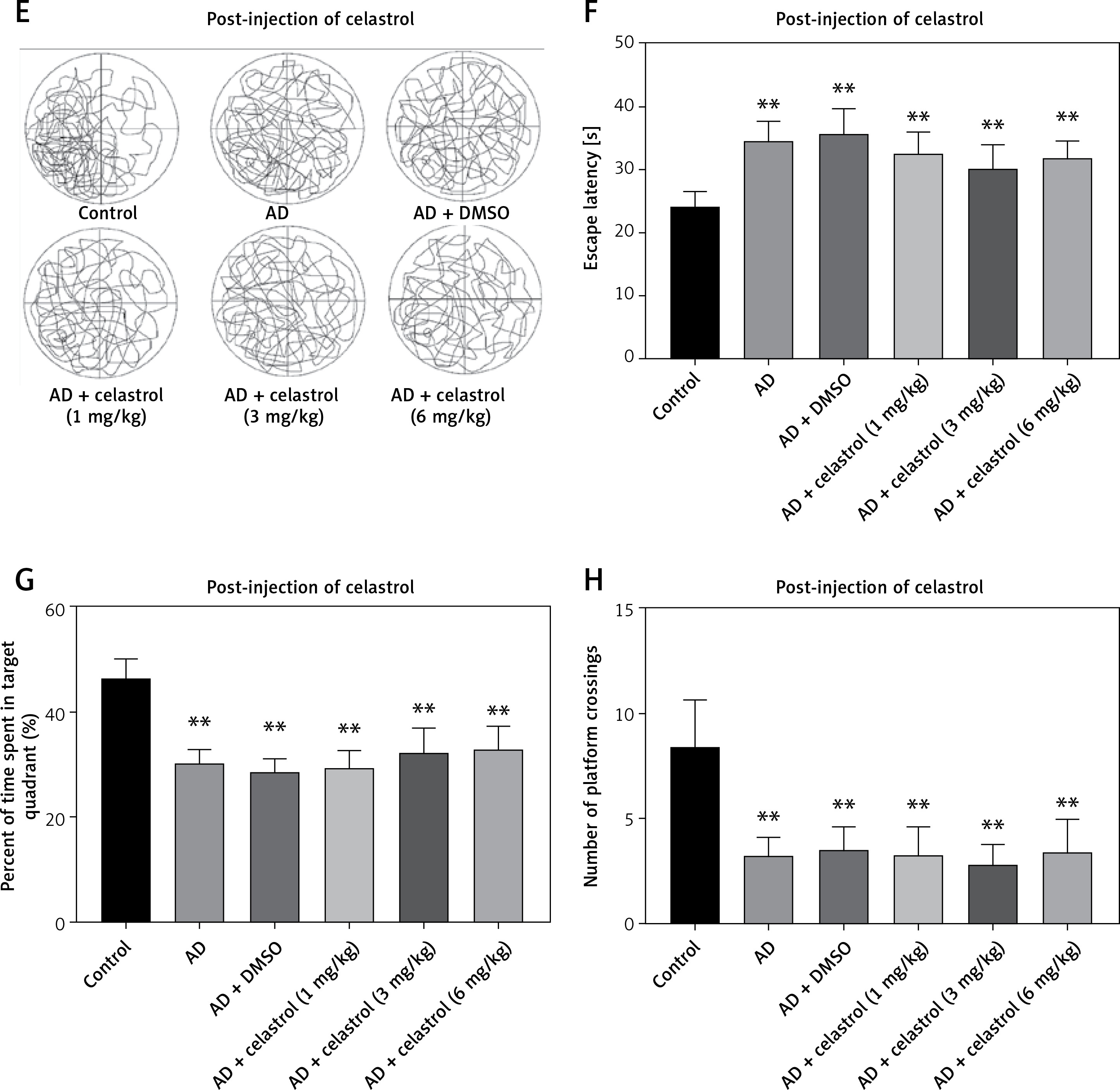
We used the Morris water maze test to evaluate the effects of celastrol on memory protection. The mice’s escape latency decreased gradually over a period of 5 days. The AD group took longer to find the hidden platform compared to the control group (Figures 2 A, B). In contrast, the AD groups treated with celastrol showed significant improvement. The swimming patterns of each group showed similar trends (Figures 2 A, B). During the probe test, the high-dose celastrol-treated AD group spent more time in the target quadrant and crossed the target platform more frequently (Figures 2 C, D). Celastrol treatment was found to enhance spatial learning and memory in AD mouse models. However, post-injection of different doses of celastrol did not significantly reverse the AD-induced impairments in escape latency, time spent in the target quadrant, or crossing frequency of the target platform (Figures 2 E, H). These results indicate that post-treatment with celastrol does not reverse the AD-induced deficits in learning and memory.
Figure 2
Celastrol pre-treatment attenuates Alzheimer’s disease (AD)-induced learning and memory impairment (Morris water maze test) in mice. A – Video tracks of the probe trial with celastrol pre-treatment. B – Escape latency to identify the hidden platform with celastrol pre-treatment. C – Percentage of time spent in the target quadrant with celastrol pre-treatment. D – Number of platform crossings with celastrol pre-treatment; **P < 0.01 compared to Control; ##P < 0.01 compared to AD + DMSO E – Video tracks of the probe trial with celastrol post-treatment. F–H Escape latency, percentage of time spent in the target quadrant and number of platform crossings with celastrol post-treatment. Error bars represent the means ± SEM of three independent experiments; **P < 0.01 compared to Control
Celastrol inhibited inflammation and oxidative stress in AD mice
Neuroinflammation could be the driving force behind the development of AD [30], leading us to explore how celastrol treatment affects inflammation and oxidative stress. Our results showed abnormal levels of inflammatory cytokines (TNF-α, IL-10, and IL-1β) in the brain tissue supernatant of AD model mice (Figures 3 A–C). Nevertheless, treatment with celastrol effectively restored these cytokine levels, demonstrating its ability to suppress inflammation in AD mice.
Figure 3
Celastrol inhibits inflammation in Alzheimer’s disease (AD) mice. A–C – tumor necrosis factor α (TNF-α), interleukin 10 (IL-10), and IL-1β levels in the supernatant of mouse brain tissues. D – hematoxylin and eosin (H&E) staining of mouse brain tissue sections. Error bars represent the means ± SEM of three independent experiments; *P < 0.05 compared to Control; **P < 0.01 compared to Control; ##P < 0.01 compared to AD + DMSO E – Nissl staining of mouse brain tissue sections
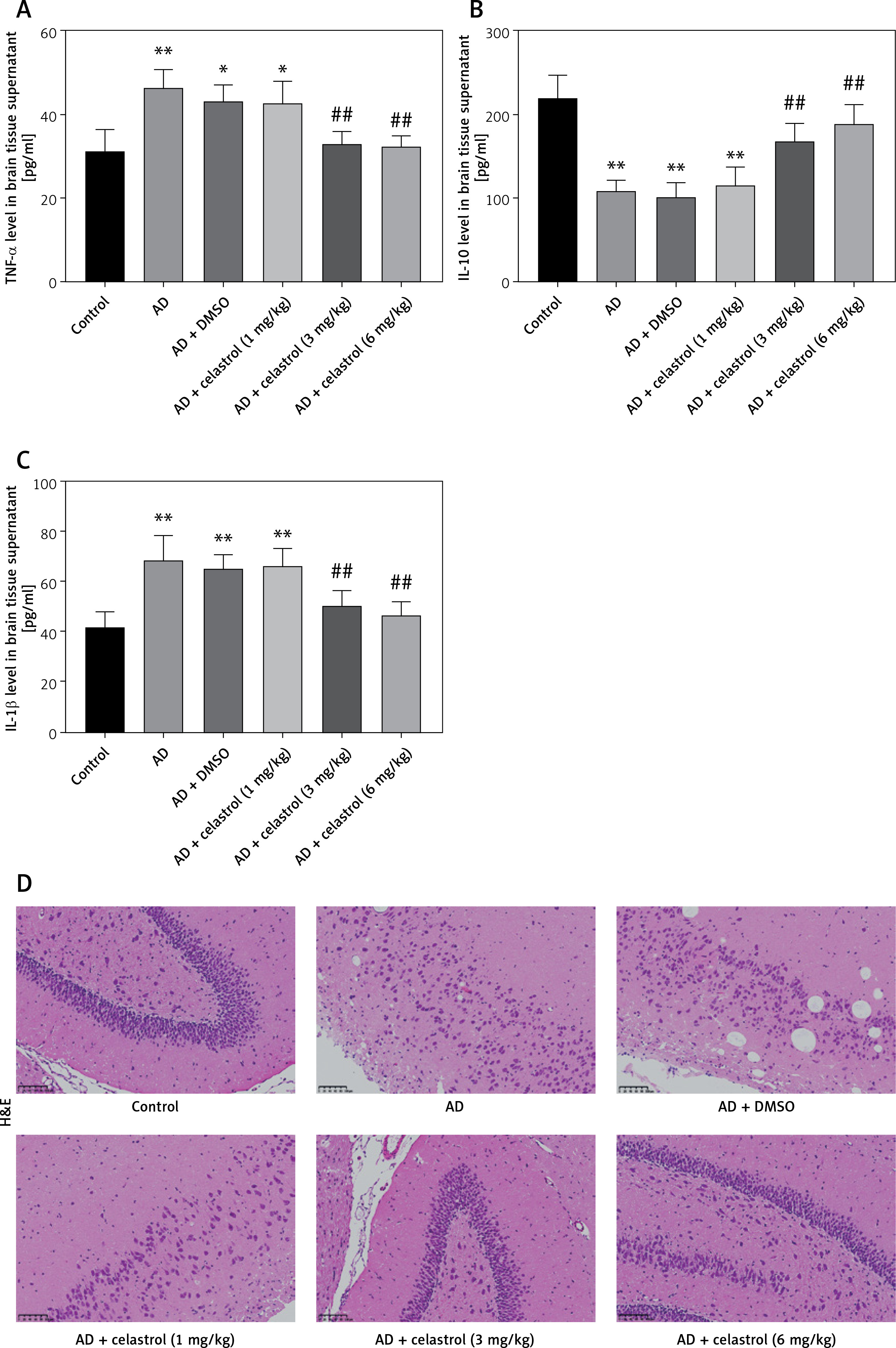
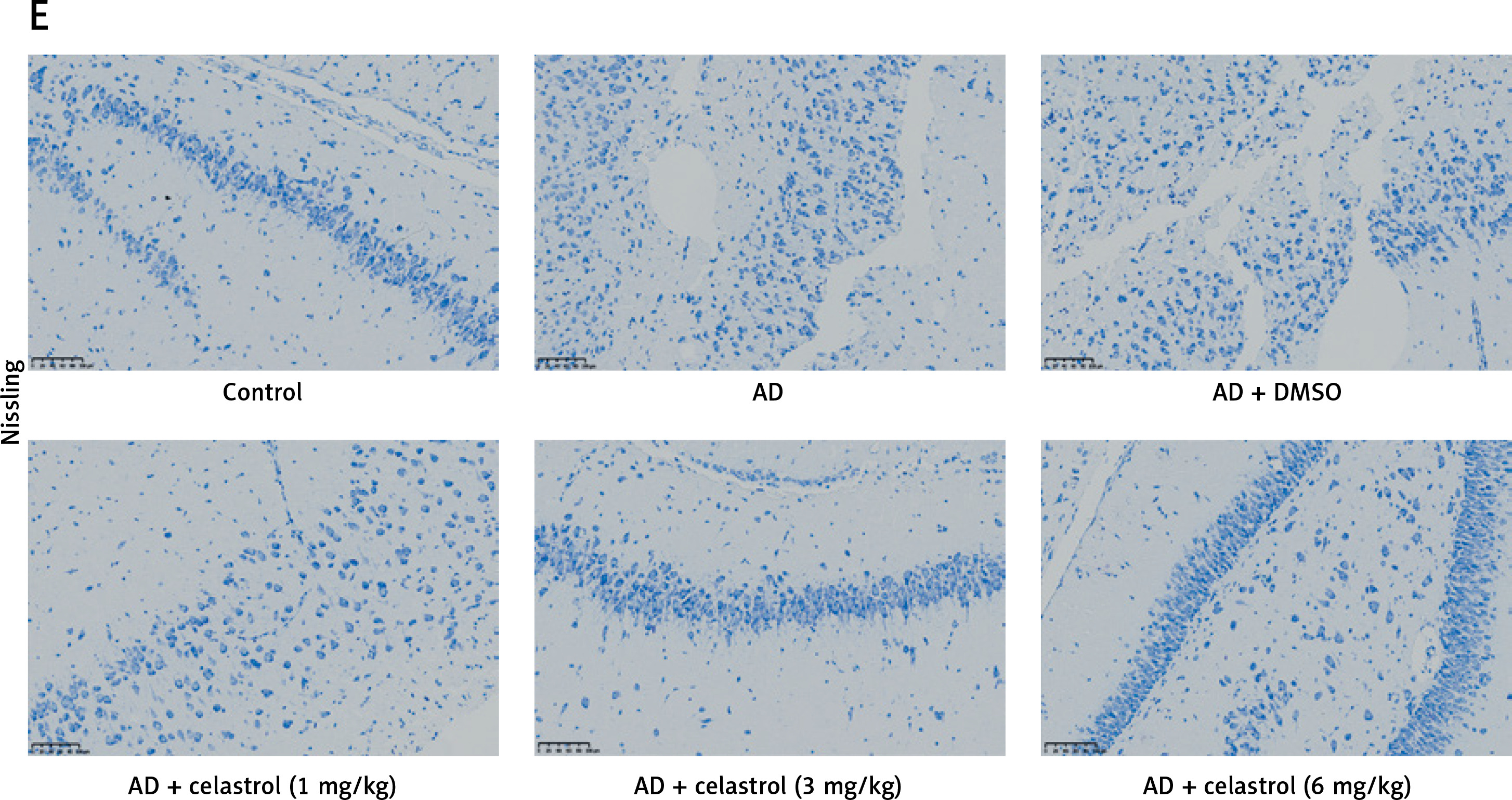
In the control group, neurons displayed numerous densely stained toluidine blue granules in the cytoplasm (Figures 3 D, E). However, in the AD group treated with celastrol, the Nissl bodies in neurons showed a significant decrease or disappearance. The histomorphological changes observed in the transgenic mice treated with different doses of celastrol varied. At 1 mg/kg, Nissl bodies in neurons showed various degrees of reduction, with lighter staining. At 3 mg/kg, neuronal atrophy and reduced Nissl bodies persisted, though less than in the vehicle-treated group. Furthermore, Nissl bodies were faintly visible in the celastrol-treated group. At 6 mg/kg, surviving neurons displayed a notable increase in Nissl bodies after celastrol treatment compared to the AD group. Ultimately, AD mice had fewer surviving neurons, and celastrol treatment could help improve neuronal survival.
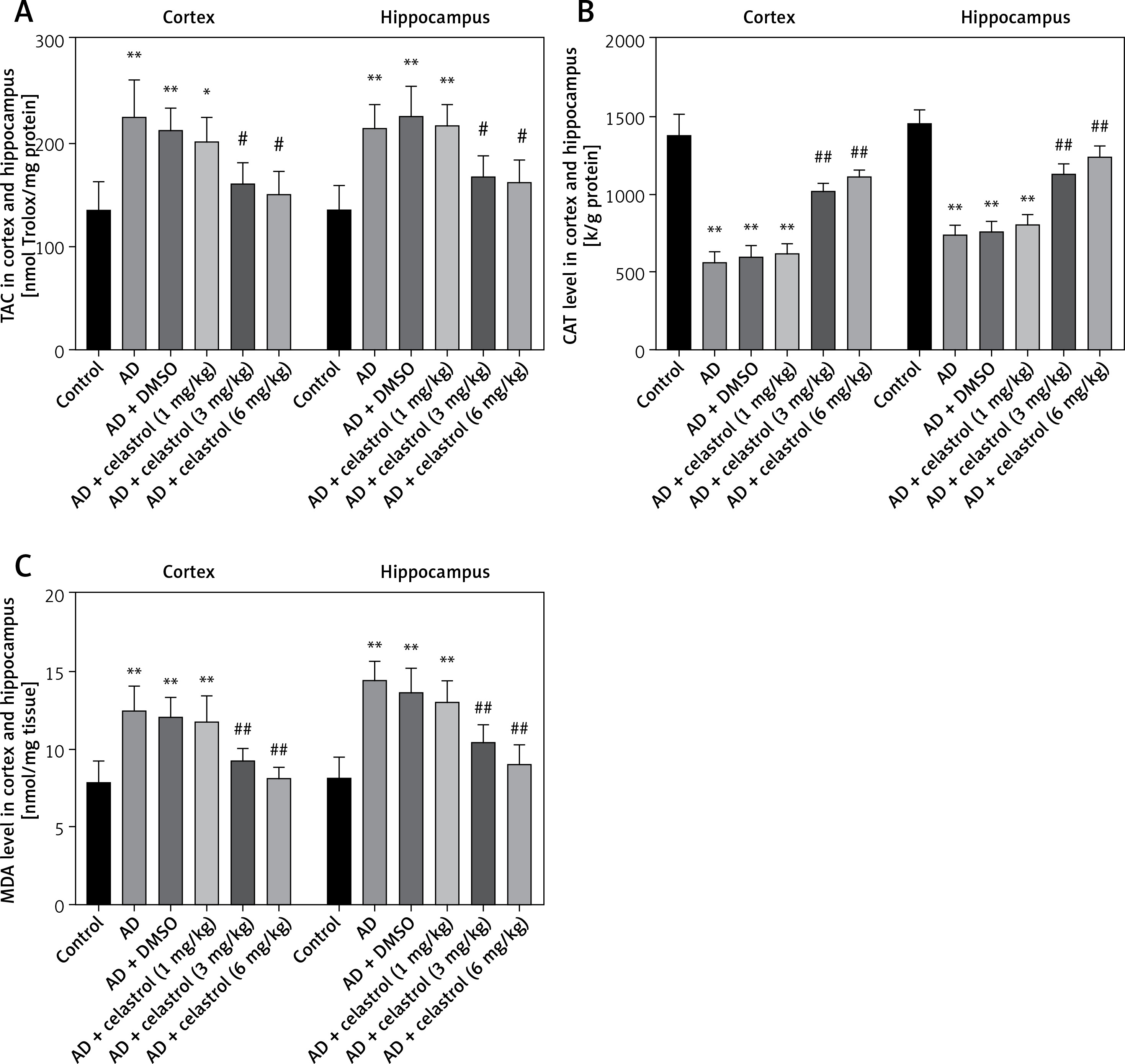
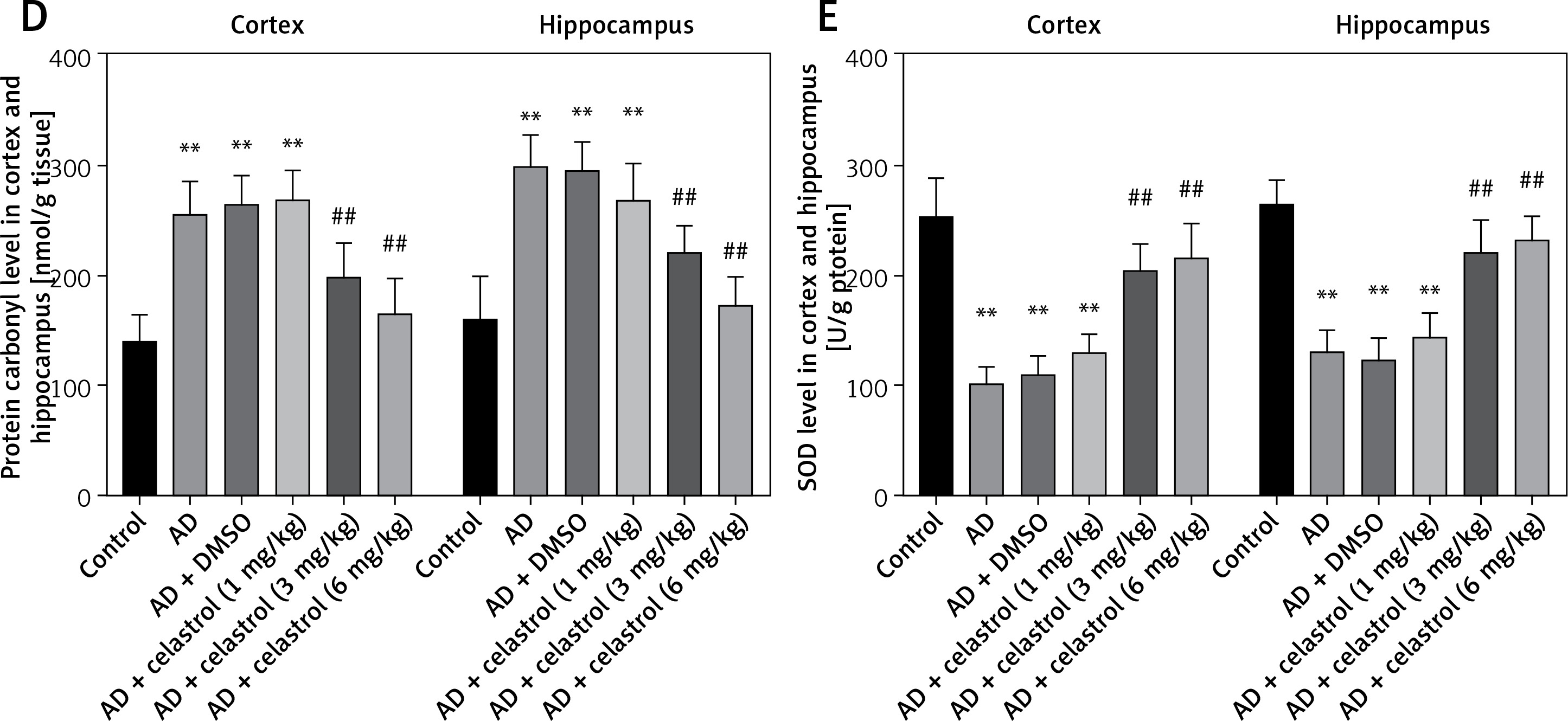
Reactive oxygen species (ROS) can be produced by endogenous sources, such as inflammation [31]. Cell defense mechanisms, including superoxide dismutase (SOD), catalase (CAT), and malondialdehyde (MDA) [32], can regulate ROS-initiated oxidative stress. In this study, the total antioxidant capacity (TAC) of the cortex and hippocampus was significantly elevated. Treatment with celastrol may inhibit the increase in TAC in the cortex and hippocampus (Figure 4 A). Additionally, the AD mouse group treated with celastrol showed lower levels of protein carbonyl and MDA and higher activity of CAT and SOD compared to the AD group (Figures 4 B–E). In conclusion, celastrol significantly inhibited inflammation and oxidative stress in AD mice.
Figure 4
Celastrol inhibits oxidative stress in AD mice. A – the TAC levels in the cortex and hippocampus; B – the CAT levels in the cortex and hippocampus; C – the MDA levels in the cortex and hippocampus. Error bars represent the means ± SEM of three independent experiments. *P < 0.05 compared to Control; **P < 0.01 compared to Control; #P < 0.01 compared to AD + DMSO; ##P < 0.01 compared to AD + DMSO. D – the protein carbonyl levels in the cortex and hippocampus; E - the SOD levels in the cortex and hippocampus. Error bars represent the means ± SEM of three independent experiments. **P < 0.01 compared to Control; ##P < 0.01 compared to AD + DMSO
TAC – total antioxidant capacity, CAT – catalase, MDA – malondialdehyde. SOD – superoxide dismutase.
Celastrol suppressed AD progression by inhibiting ER stress in AD mice
We conducted western blot analysis to evaluate the protein expression of markers associated with Alzheimer’s disease, including beta-site APP-cleaving enzyme 1 (BACE1), low-density lipoprotein receptor-related protein 1 (LRP1), neprilysin (NEP), and receptor for advanced glycation end products (RAGE). Our results showed significant differences in marker expression between healthy mice and AD model mice. However, celastrol treatment effectively restored protein expression in the AD group (Figures 5 A, B). Furthermore, we obtained brain tissues from each group and used immunohistochemistry to visualize BACE1 expression in these tissues. We found that BACE1 was highly expressed in AD mice, but celastrol treatment appeared to inhibit its expression (Figures 6 A, C). In conclusion, our findings suggest that celastrol treatment may inhibit the progression of Alzheimer’s disease in vivo.
Figure 5
Celastrol suppresses Alzheimer’s disease (AD) progression by inhibiting ER stress in AD mice (Western blot). A – Protein levels of AD-related markers (beta-site APP-cleaving enzyme 1 (BACE1), low-density lipoprotein receptor-related protein 1 (LRP1), neprilysin (NEP), and receptor for advanced glycation end products (RAGE)) and endoplasmic reticulum (ER) stress-related markers (caspase-12, C/EBP homologous protein (CHOP), and glucose-regulated protein 78 (GRP78)) in the brain tissues of mice B–C – Protein levels of AD-related markers (beta-site APP-cleaving enzyme 1 (BACE1), low-density lipoprotein receptor-related protein 1 (LRP1), neprilysin (NEP), and receptor for advanced glycation end products (RAGE)) and endoplasmic reticulum (ER) stress-related markers (caspase-12, C/EBP homologous protein (CHOP), and glucose-regulated protein 78 (GRP78)) in the brain tissues of mice. Error bars represent the means ± SEM of three independent experiments; **P < 0.01 compared to Control; ##P < 0.01 compared to AD + DMSO
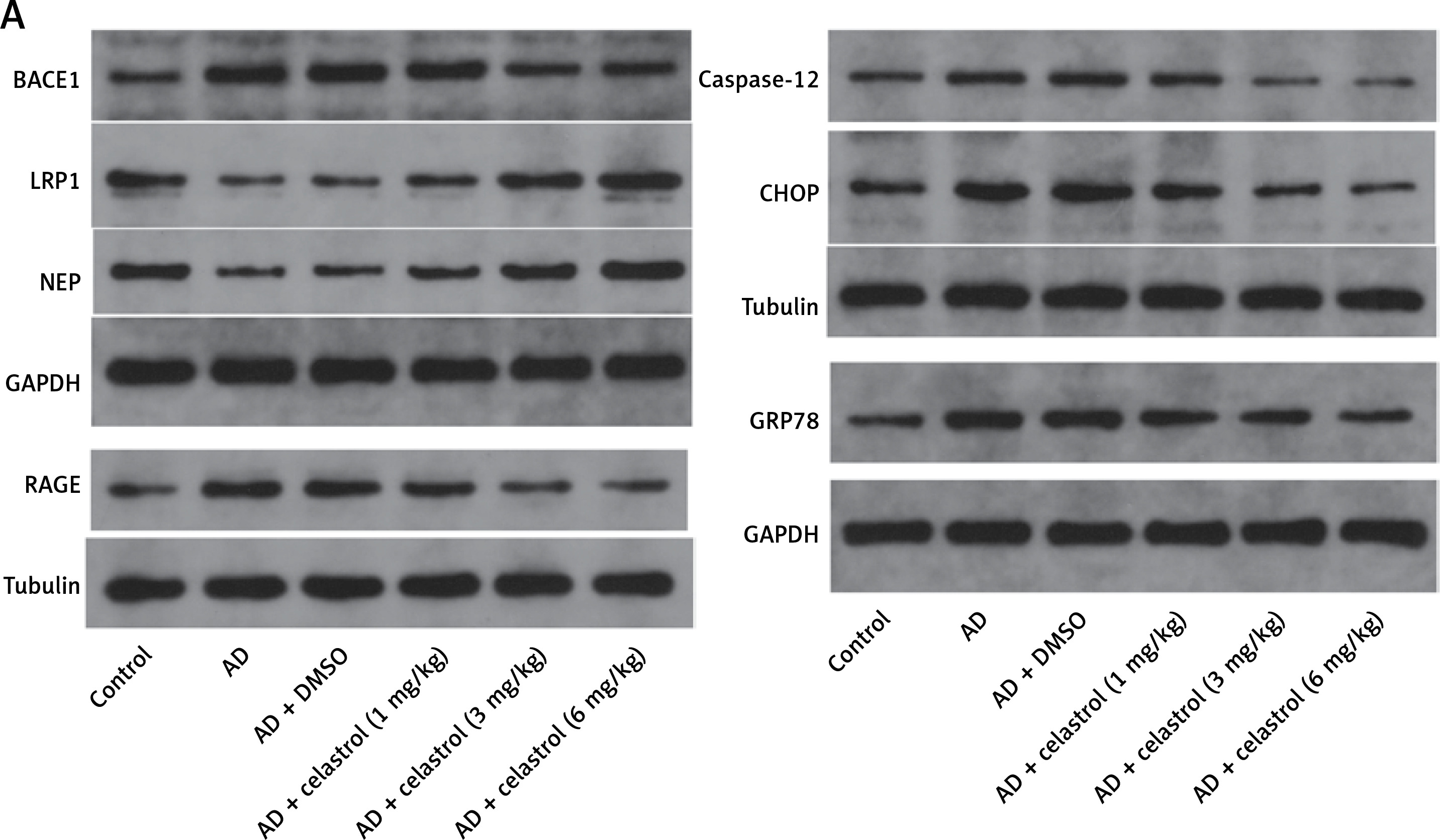
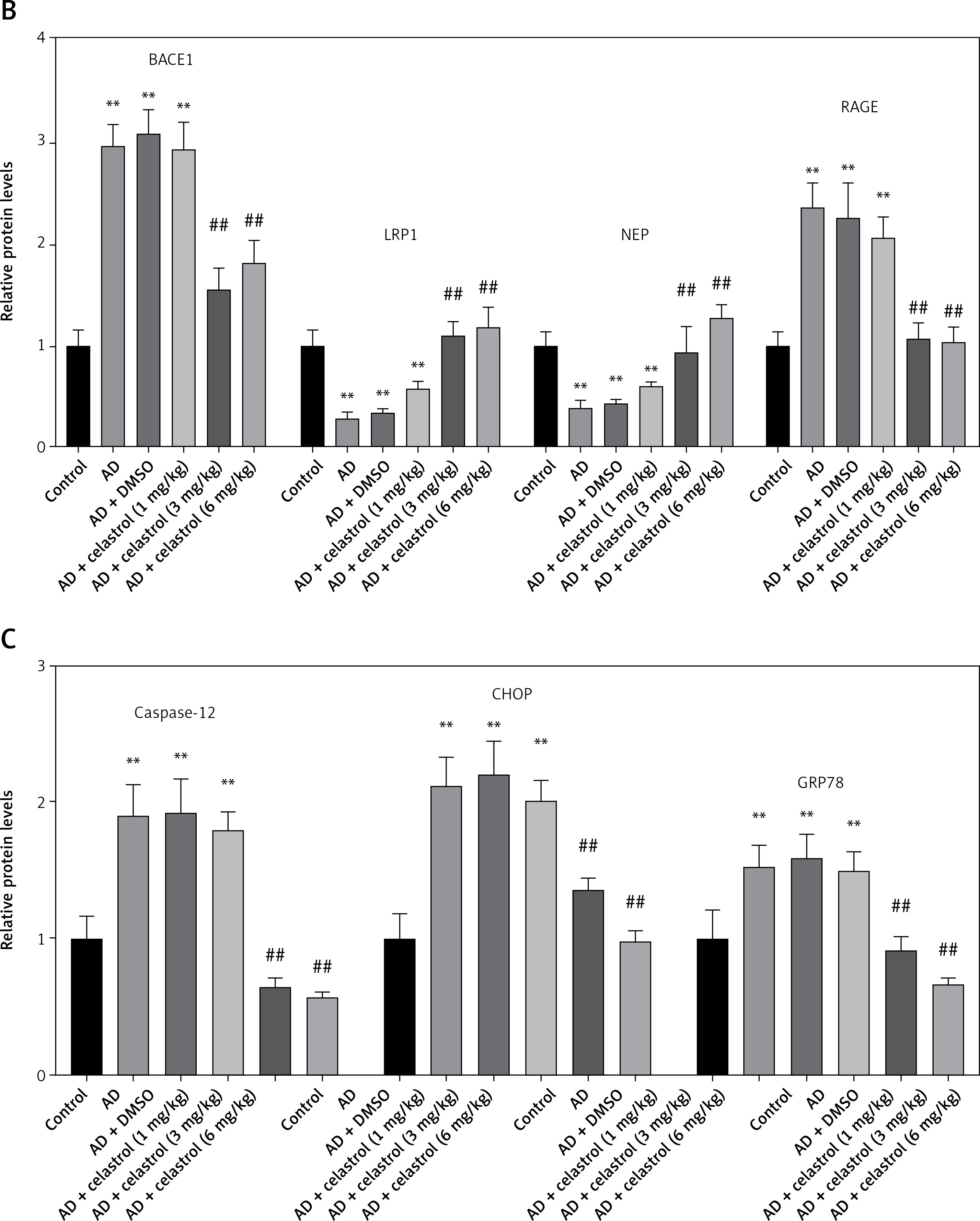
Figure 6
Celastrol suppresses Alzheimer’s disease (AD) progression by inhibiting endoplasmic reticulum (ER) stress in AD mice (immunohistochemistry). A–C – Immunohistochemical staining of beta-site APP-cleaving enzyme 1 (BACE1) or glucose-regulated protein 78 (GRP78) in brain tissues of mice. Error bars represent the means ± SEM of three independent experiments; **P < 0.01 compared to Control; ##P < 0.01 compared to AD + DMSO
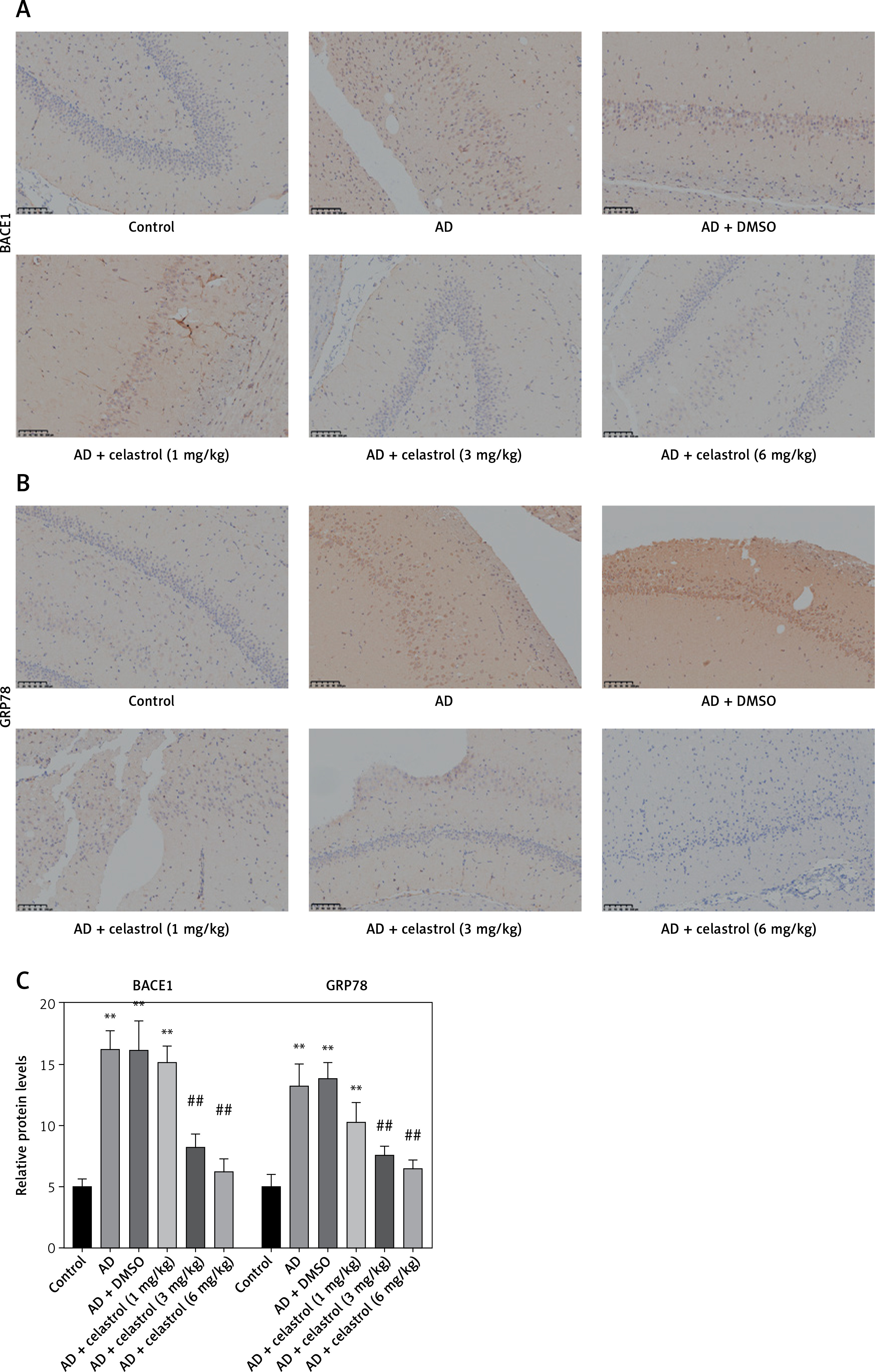
ER is a ubiquitous organelle present in eukaryotic cells [33, 34]. Disruption of ER homeostasis by physiological and pathological factors, including depletion of calcium and disturbances in lipid synthesis, can lead to cellular dysfunction [35, 36]. These conditions, collectively referred to as conformational anomalies within ER stress, contribute to cellular damage in Alzheimer’s disease [37]. To investigate the effects of celastrol on ER stress, we examined the expression of ER stress-associated genes. The results of a western blot assay indicated that the protein expression levels of caspase-12, CHOP, and GRP78 were significantly lower in the celastrol-treated AD group compared to the AD group (Figures 5 A, C). Immunohistochemical analysis also supported these results (Figures 6 B, C). Overall, treatment with celastrol may suppress the progression of AD by inhibiting ER stress in AD mice.
Celastrol attenuated AD-induced learning and memory impairment by inhibiting ER stress-induced inflammation and oxidative stress in AD mice
In order to explore the role of ER stress in AD progression, some mice were injected intraperitoneally with tunicamycin (an ER stress activator) or TUDCA (an ER stress inhibitor). In the Barnes maze cognitive test, AD mice took longer to identify the goal box after a training period. Treatment with celastrol or TUDCA attenuated the AD-induced effect (Figures 7 A, B). Moreover, simultaneous intraperitoneal injection of celastrol and tunicamycin partially restored the inhibitory effect of celastrol treatment on AD-induced learning and memory impairment. Additionally, western blotting analysis of ER stress-associated genes (caspase-12, CHOP, and GRP78) showed that celastrol or TUDCA treatment inhibited AD-induced ER stress. Tunicamycin treatment was able to partly negate the effects of celastrol treatment on ER stress in AD mice (Figure 7 C). In conclusion, celastrol attenuated AD-induced learning and memory impairment by inhibiting ER stress in AD mice.
Figure 7
Celastrol attenuates Alzheimer’s disease (AD)-induced learning and memory impairment by inhibiting endoplasmic reticulum (ER) stress-induced inflammation in AD mice. A – Training performance during the Barnes maze cognitive test. B – Memory phase performance during the Barnes maze cognitive test. C – Protein levels of ER stress-related markers (caspase-12, CHOP, and glucose-regulated protein 78 (GRP78)) in the brain tissues of mice. D–F – tumor necrosis factor α (TNF-α), interleukin 10 (IL-10), and IL-1β levels in the supernatant of brain tissues. Error bars represent the means ± SEM of three independent experiments; **P < 0.01 compared to Control; ##P < 0.01 compared to AD; &&P < 0.01 compared to AD + celastrol (3 mg/kg)
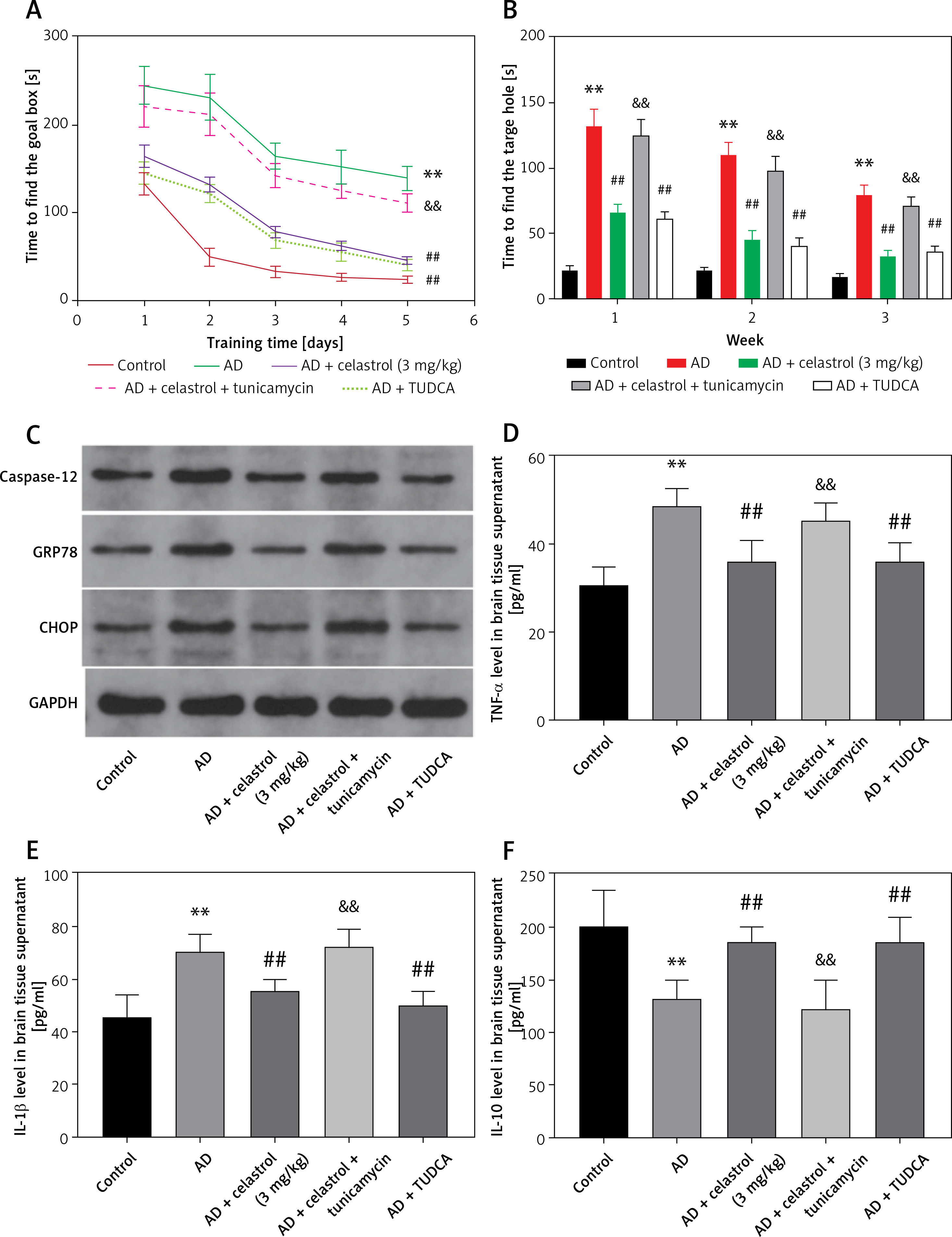
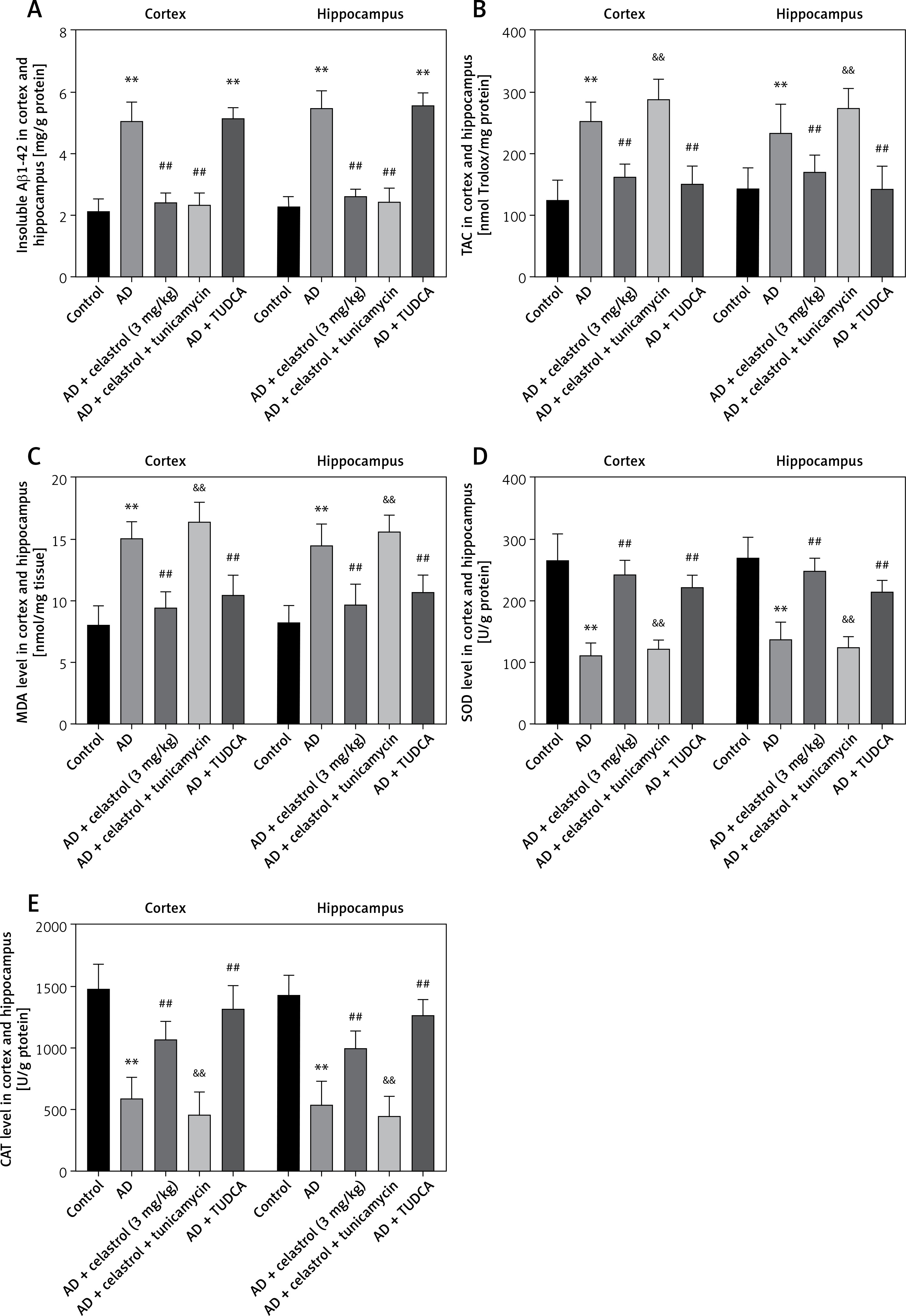
Next, we evaluated the effects of ER stress on inflammation and oxidative stress during the progression of Alzheimer’s disease. Treatment with celastrol and TUDCA resulted in a suppression of inflammatory cytokine levels (TNF-α, IL-10, and IL-1β) in AD mouse models. Additionally, the ER stress activator tunicamycin was able to reverse the inhibitory effects of celastrol treatment on AD-induced inflammation (Figures 7 D–F). Celastrol significantly reduced the level of insoluble Aβ1-42 in the brain, which was not affected by tunicamycin or TUDCA treatment (Figure 8 A), indicating that tunicamycin or TUDCA did not clear insoluble Aβ1-42. Furthermore, treatment with celastrol or TUDCA led to a decrease in levels of MDA and TAC in the cortex and hippocampus, while also increasing CAT and SOD activity (Figure 8 B–E). However, tunicamycin treatment partially restored the effects of celastrol treatment on oxidative stress in AD mice.
Figure 8
Celastrol attenuates Alzheimer’s disease (AD)-induced learning and memory impairment by inhibiting ER stress-induced oxidative stress in AD mice. A–E – Insoluble Aβ1-42, total antioxidant capacity (TAC), catalase (CAT), malondialdehyde (MDA), and superoxide dismutase (SOD) levels in the cortex and hippocampus. Error bars represent the means ± SEM of three independent experiments; **P < 0.01 compared to Control; ##P < 0.01 compared to AD; &&P < 0.01 compared to AD + celastrol (3 mg/kg)
Discussion
In this study, it was revealed that pre-treatment with celastrol, as opposed to post-treatment, significantly attenuated AD-induced learning and memory impairment in mice. Pre-treatment with celastrol inhibited inflammation and oxidative stress in the AD mouse model. Additionally, celastrol suppressed AD progression by inhibiting ER stress in the AD mouse model. These findings enhance our understanding of how celastrol affects learning and memory deficits, potentially leading to the development of novel neuroprotective treatment strategies. It is worth noting that only pre-treatment with celastrol has an anti-AD effect, and several mechanisms may contribute to this. Firstly, administering celastrol after AD-induced injury has occurred may limit its ability to impact the underlying mechanisms or pathways that drive the process. Celastrol may be less effective at reversing or modifying the biological changes in the brain that have already taken place. Secondly, administering celastrol before the AD-induced injury reaches a critical stage may prevent or delay the development of pathological processes, making the treatment more effective at maintaining neurophysiological functions. Lastly, administering celastrol before the onset of AD may allow it to interact with its target more effectively, enhancing its efficacy in preventing AD-induced injury.
According to a recent survey, AD affects approximately 10% of elderly people (> 65 years old) [38]. However, the medications currently available for AD only partly reduce the symptoms and often come with adverse effects. This underscores the urgent need for novel and effective treatments [39]. Therefore, exploring additional AD-related biomarkers holds significant potential for future research. In our study, we focused on inflammation, oxidative stress, and ER stress as potential AD-related biomarkers. Our findings suggest that celastrol, a natural compound derived from traditional Chinese medicine, has the potential to attenuate learning and memory impairment in Alzheimer’s disease. This indicates that targeting ER stress-induced inflammation and oxidative stress pathways may represent a promising therapeutic approach for treating AD.
Celastrol is primarily extracted from Celastraceae and possesses several protective functions [40]. Research has shown that celastrol can reduce cell death and Aβ production in cell experiments [41]. Therefore, we investigated its protective effects in Aβ25-35-induced AD model mice and found that celastrol mitigated Aβ-induced learning and memory deficits.
Celastrol has demonstrated antioxidant, anti-inflammatory, and anticancer activities in various experimental settings [14, 42, 43]. In this study, it was observed that celastrol treatment significantly restored the expression of BACE1, LRP1, NEP, and RAGE in the brain. BACE1 is considered a potential target as it is involved in the production of toxic Aβ42 peptides, leading to amyloid plaque deposits and ultimately causing AD [44]. Celastrol’s inhibition of HACE1 may be a significant factor in its effect on AD. LRP1 regulates Aβ metabolism and helps maintain brain homeostasis [45]. Activation of LRP1 by celastrol may lead to the attenuation of AD symptoms. NEP is a major catabolic enzyme of Aβ, and increasing its expression has been shown to have beneficial effects in animal models of Alzheimer’s disease [46–48]. Celastrol may promote the clearance of Aβ by activating NEP. RAGE is another important protein that can worsen the progression of AD. Certain drugs can alleviate AD by regulating RAGE/NOX4-mediated oxidative stress and inflammation [49], which may also play a role in celastrol’s effectiveness. Our study corroborated these findings, demonstrating the potential of celastrol to inhibit inflammation and oxidative stress while suppressing ER stress-induced damage in AD mice. Celastrol’s ability to modulate multiple pathways involved in AD pathophysiology, including endoplasmic reticulum stress, inflammation, and oxidative stress, highlights the potential benefits of a multi-target approach in treating complex neurodegenerative diseases such as AD.
ER stress is central to neuronal damage mechanisms, particularly apoptosis, implicated in AD onset [37]. The unfolded protein response (UPR), initiated in response to ER stress, regulates neuronal function and has been implicated in brain injuries [50, 51]. Misfolded protein accumulation and subsequent ER stress can lead to neuronal dysfunction in neurodegenerative diseases, with the surrounding glial cells contributing to abnormal inflammatory signaling and disease progression. Studies have reported on UPR upregulation in AD brain samples [52], suggesting its involvement in neurodegeneration, possibly through Aβ accumulation [53]. We investigated the regulatory and functional aspects of ER stress in AD model mice. Caspase-12, CHOP, and GRP78 are three major ER stress-related markers [54]. The results showed that celastrol suppresses AD progression by cleaving these ER stress-related proteins in AD mice, suggesting the potential of celastrol to suppress AD progression by inhibiting ER stress, thereby attenuating AD-induced learning and memory impairment. Understanding the mechanism by which ER stress specifically contributes to neuronal damage in AD is paramount for advancing its clinical application. Our findings shed light on the pivotal role of ER stress in AD pathogenesis, offering insights into potential clinical interventions.
In conclusion, celastrol may confer protective effects against learning and memory decline through its anti-inflammatory and anti-oxidative stress properties in AD model mice. The study provides mechanistic insights into the pathogenesis of Alzheimer’s disease, specifically the role of endoplasmic reticulum stress, inflammation, and oxidative stress in disease progression. Understanding these underlying mechanisms is crucial for developing targeted therapies and interventions for AD. Moreover, the identification of celastrol as a potential therapeutic agent for AD offers an alternative treatment option beyond conventional pharmaceuticals. Natural compounds like celastrol may provide new avenues for drug discovery and development in the field of AD research. Understanding the molecular mechanisms underlying celastrol’s therapeutic effects in AD may pave the way for personalized medicine approaches in Alzheimer’s disease treatment.
While the present study presents promising findings, there are certain limitations that should be considered. First, the animal model of AD may not fully replicate the complex pathophysiology and progression of the disease in humans. Translating findings from animal studies to clinical applications in humans can be challenging. Second, the applicability of our findings to human patients with Alzheimer’s disease requires further validation through clinical trials and human studies. Third, while celastrol shows promise, its specificity, bioavailability, potential side effects, and long-term safety profile need further investigation. Further research is needed to validate the therapeutic potential of celastrol in Alzheimer’s disease and address the gaps and uncertainties in understanding its mechanisms of action and clinical implications.