Introduction
The relative scarcity of information concerning tubular, interstitial, and vascular changes has been the target of criticism in terms of the current classifications of lupus nephritis (LN) because of the larger amount of information available regarding glomerular changes, even though their potential for independent progression is known. The International Society of Nephrology/Renal Pathology Society (ISN/RPS) 2003 classification of LN [1] has been widely accepted, has high intra-observer and inter-observer concordance, and can be used to guide therapeutic strategies and provide prognostic information. However, this classification is not based on the pathophysiology of the underlying disease. Some additional lesions that contribute to the presentation of LN (Table I) are somewhat less valued according to the current classification; however, because of their influence on the prognosis of disease, they should be recognised and valued [2].
Table I
Forms of renal involvement in SLE
So far, few efforts have been made in this direction. In 2015, Wilhelmus et al. discussed the necessity to re-evaluate the histological lesions (including lupus podocytopathy, collapsing glomerulopathy, thrombotic microangiopathy, tubulointerstitial, and vascular lesions) [3]. Moreover, in 2018, as result of the International Nephropathology Working Group Consensus, a revision of the ISN/RPS classification for LN was published to reach a consensus on recently raised issues concerning problems with definitions of lupus nephritis lesions, including tubulointerstitial, vascular, and podocyte lesions [4].
This study reviewed the importance of lesions that are less recognised by the current classification and focused on tubulointerstitial, vascular, and podocyte involvement not included in the original classification. This is timely because the aforementioned involvement has increasingly been recognised as important in the pathogenesis and prognosis of LN, as will be presented in this review.
Tubulointerstitial nephritis
The current LN classifications emphasise glomerular inflammation and scarring. However, tubulointerstitial inflammation and fibrosis are better predictors of progression to kidney insufficiency. In fact, the importance of tubulointerstitial injury and damage in short-term and long-term prognosis has been repeatedly indicated in the literature [5–11].
Tubulointerstitial involvement is a well-recognised characteristic of LN that occurs in 66% of renal biopsy samples from patients with systemic lupus erythematosus (SLE) [12], which is mostly represented by lesions with no clinical significance, such as droplets of reabsorption of cytoplasmic proteins, lipid vacuoles, and tissue antinuclear antibodies (“tissue ANA”) [13] (Figure 1 A). In the majority of tubulointerstitial lesion cases, tubular atrophy associated with interstitial fibrosis and inflammation accompanies severe glomerular lesions [14]. However, the predominant or isolated presence of tubulointerstitial changes in the framework of minimal or absent tubulointerstitial abnormalities in patients with SLE is rare. Only 15 cases have been reported [15] thus far, and the immunopathogenesis of predominantly tubulointerstitial LN remains unknown.
Figure 1
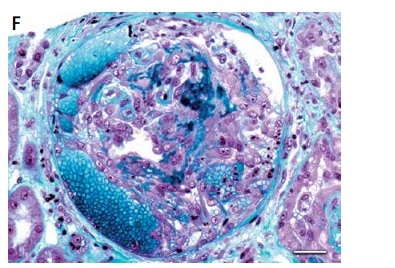
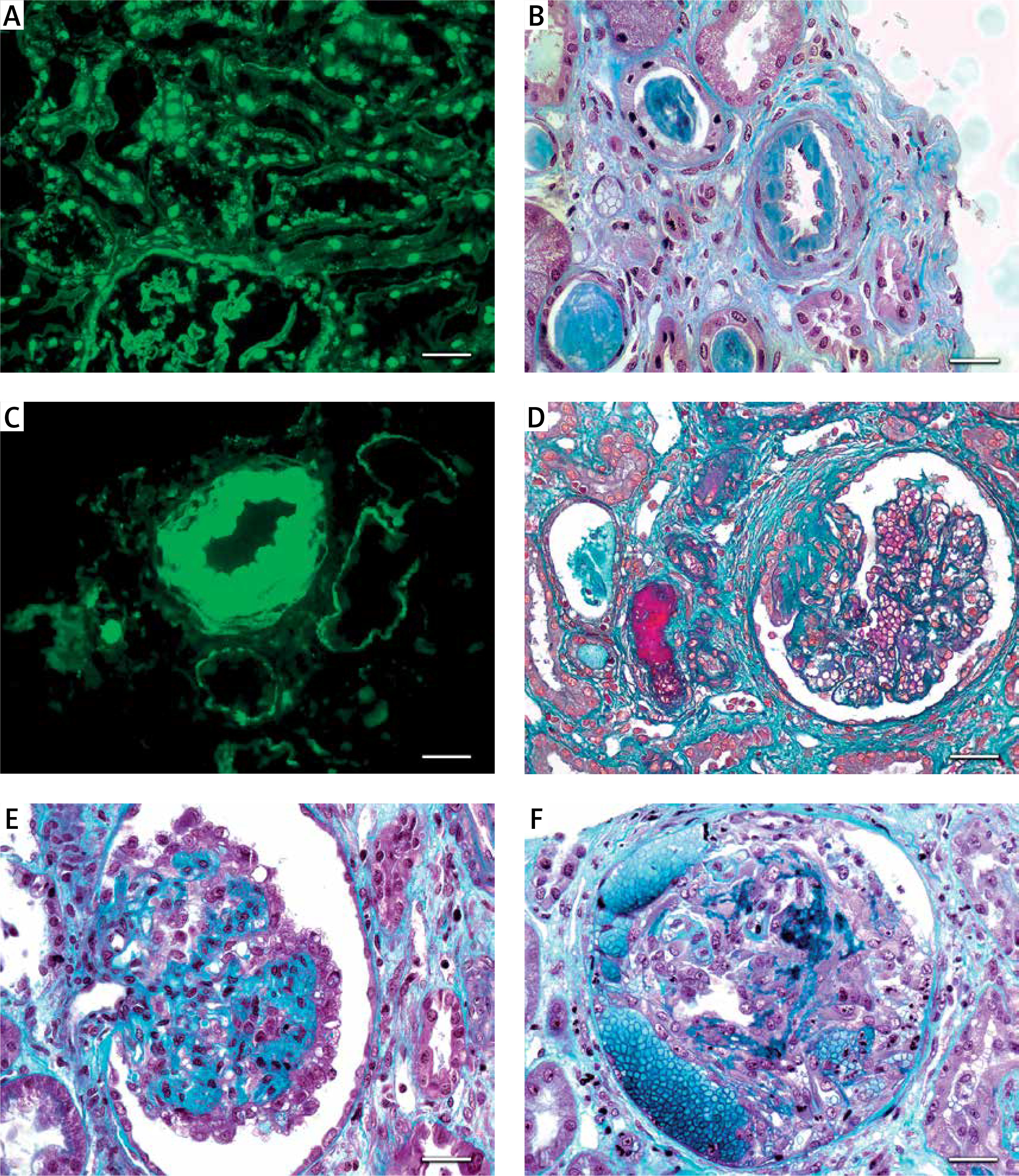
Lupus nephritis. A – Tissue antinuclear antibodies (ANA). The immunofluorescence micrograph shows tubular ANA reactivity in this cryostat section stained for IgG, referred to as “tissue ANA’ (bar = 15 μm). B – Non-inflammatory necrotising vasculopathy. The lumen of an arteriole is narrowed by intimal hyaline deposits without inflammation of the vessel wall (Masson’s trichrome; bar = 30 μm). C – Non-inflammatory necrotising vasculopathy. The immunofluorescence micrograph shows intense staining for C1q within the intima and media of an arteriole (bar = 30 μm). D – Thrombotic microangiopathy. The glomeruli show proliferative class changes, and the arteriole shows total occlusion by intraluminal accumulation of fibrin and erythrocytes (Masson’s trichrome; bar = 15 μm). E – Focal segmental glomerulosclerosis (FSGS; collapsing variant). In the collapsed tuft, the glomerular basement membranes are imploded without an appreciable increase in the matrix material. The podocytes overlying the collapsed tuft are markedly hyperplastic with enlarged vesicular nuclei (Masson’s trichrome; bar = 30 μm). F – Lupus nephritis class IV with antineutrophil cytoplasmic antibody (ANCA)-associated necrotising and crescentic glomerulonephritis. A glomerulus shows complete fibrinoid necrosis associated with a circumferential cellular crescent and haemorrhage (Masson’s trichrome; bar = 30 μm)
However, the absence of significant glomerular involvement implies that circulating immune reagents specifically interact with one or more tubulointerstitial autoantigens, which are missing or not expressed in the glomeruli. Furthermore, the underlying mechanism seems to be in situ formation of immune complexes after the binding of circulating autoantibodies to exogenous or native antigens [16].
It has become clear that the immunologic characteristics and probable underlying mechanisms are vastly different between lupus glomerulonephritis and tubulointerstitial inflammation (TII) at the time of biopsy. In lupus glomerulonephritis, the degree and type of renal involvement vary with the ISN/RPS class. Proliferative forms of LN (classes III and IV) are characterised by inflammation. Active glomerular lesions feature prominent subendothelial immune complexes that can fill the glomerular capillary loops (hyaline thrombi). T cells and macrophages accumulate at the sites of subendothelial immune complexes and lead to the breakup of the glomerular basal membrane, fibrinoid necrosis, and cellular crescents. Neutrophils are not prominent in glomerular capillaries, except when fibrinoid necrosis and crescent formation occur. B cells and plasma cells are rare in glomeruli and are usually confined to the intravascular space [11].
However, although lupus glomerulonephritis is a manifestation of systemic autoimmunity, TII is associated with local networks of adaptive immune cells in situ with predictive amplification of local inflammation and tissue damage. In addition, ill-defined networks of innate and effector immune cells probably contribute to the severity of local inflammation. It has been demonstrated that tubulointerstitial infiltration by B and T lymphocytes is common in LN. In a cohort of 68 lupus nephritis biopsies, more than half of the samples showed that the tubulointerstitial infiltrate was organised into well-circumscribed T:B cell aggregates or germinal centres (GCs) containing follicular dendritic cells. Sampling of the in situ-expressed immunoglobulin repertoire revealed that both histological patterns were associated with intrarenal B cell clonal expansion and ongoing somatic hypermutation. However, in the GC histology, the proliferating cells were CD138−CD20+ centroblasts, whereas they were CD138+CD20low/− plasmablasts in T:B aggregates. The presence of GCs or T:B aggregates was strongly associated with tubular basement membrane immune complexes. These data implicate tertiary lymphoid neogenesis in the pathogenesis of lupus TII [17, 18].
The autoantigen driving in situ B-cell selection in TII seems to be vimentin, a major constituent of the intermediate filament family of proteins that might be expected to be very immunogenic. Vimentin is highly expressed in activated T cells and macrophages [19–21] and also in TII [19]. Moreover, vimentin can bind Dectin-1, a C-type lectin receptor expressed on dendritic cells, macrophages, and B cells [22]. Therefore, vimentin might be an immunodominant pattern of inflammation [11].
These observations suggest that, in addition to the systemic autoimmune processes that have been implicated in glomerulonephritis, the local immune responses in situ can be associated with interstitial nephritis; furthermore, it can be inferred that the assessment of interstitial inflammation captures an important pathological process that can be tissue-specific and independent of glomerular disease [11].
Proteinuria, immune complex deposition in the interstitium, proinflammatory molecules on renal tubular cells, rupture of the Bowman’s capsule, and cryptic antigen presentation by juxtaglomerular cells are some of the insults resulting in interstitial infiltration by inflammatory cells and, ultimately, tubular atrophy, collectively constituting a strong rationale for inclusion of the tubulointerstitial compartment in classification sets, prognostic markers, and outcome measures [23]. Several studies in the immunosuppressive era of lupus nephritis treatment have indicated that tubulointerstitial inflammation is prognostically more meaningful than glomerular inflammation and more likely to be correlated with elevated creatinine at time of biopsy and with risk for subsequent renal failure [9, 24–26]. Many of these studies noted that more active TII tended to be associated with active GN. However, multivariate analysis demonstrated that TII was an independent predictor of progression to renal failure [9] and correlated with serum creatinine at time of biopsy [9, 24].
According to current recommendations by ISN/RPS 2003, tubular atrophy, interstitial inflammation, and fibrosis have to be reported in the diagnostic line and graded as mild, moderate, or severe [1]. No cut-off values for this grading system are provided. It is also unclear if all three parameters should be graded separately or could be combined into one grade for tubulointerstitial damage, because interstitial fibrosis and tubular atrophy have been shown to correlate with tubulointerstitial inflammation in LN [9]. Thus, modifications to the vague points of the current classification are necessary, in order to systematise the evaluation of the tubulointerstitium so that better parameters can be evaluated to determine therapeutics and prognosis.
For this reason, a revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis was presented, with the intention of clarifying the definitions and modifying the National Institutes of Health activity and chronicity indices [4]. According to this review, this deficiency should be addressed by the development of a valid activity and chronicity index that scores the severity of tubulointerstitial injury. It proposed the gathering of data on interstitial fibrosis and tubular atrophy and interstitial infiltrates in a semiquantitative fashion in the future, rounding fibrosis to the nearest 10%, with minimal fibrosis stated as 5%. These values could then be translated into reproducible scoring categories (e.g. on a scale from 0 to 3+) based on cut-off values to be evaluated for prognostic significance, as currently done for the Banff and Oxford classifications. The review also highlighted that it is important to determine in the future whether interstitial fibrosis and tubular atrophy should be considered separately or combined into a single parameter and whether making a distinction between interstitial inflammation in areas with or without interstitial fibrosis has clinical significance.
Vascular lesions
Renal vascular complications are commonly found in SLE, and their occurrence can have a profound effect on the clinical course and choice of therapy. Although renal vascular complications have been described in several studies [27–35], they were not included in the activity and chronicity scores of the LN classification created by the National Institutes of Health. The ISN/RPS 2003 classification of LN suggests the inclusion of vascular disease only in the form of a descriptive report of the kidney biopsy results; therefore, it can be easily omitted by the pathologist [13].
This possibility of omission allowed by the ISN/RPS 2003 causes confusion for researchers of renal involvement in SLE, especially because they recognise the prognostic value of the vascular compartment in other renal pathologies, such as during the assessment of renal transplantation donors. For example, in cases of renal transplantation, the presence of vascular damage, mainly in the fibrous thickening of the arterial intima, is a reliable prognostic factor for the survival of the transplanted kidney [36, 37]. Therefore, it is still controversial whether renal vascular complications should be considered independent renal prognostic factors for SLE. A series of studies indicated that renal vascular complications are closely associated with clinical disease, disease activity, and renal outcomes, and that renal thrombotic microangiopathy (TMA) is an independent risk factor for the renal prognosis of patients with long-term LN [34, 38–41].
Renal vascular complications in LN were initially classified in 1994 by Appel et al. [27] and include the following:
Arterial sclerosis. This is defined as thickening of the intimal middle layers of the interstitial arteries and/or arteriolar hyalinosis. It is not specific for SLE and is common in patients with a history of chronic LN of any class. It is associated with aging, hypertension, and immunosuppressive drug use, and it is a relevant prognostic factor.
Uncomplicated vascular deposits of immune complexes. The most common renal vascular lesion in LN is the deposition of immune complexes in the walls of arterioles and small arteries. It is characteristic of class III and class IV, but it can also occur in class II and class V. On immunofluorescence, vascular deposits have a “full house” pattern and are positive for serum C1q. Unlike arterial sclerosis, the deposition occurs mainly in the subendothelial region. It is highly characteristic of LN (Figures 1 B, C).
Non-inflammatory necrotising vasculopathy. This is defined as necrotising alterations of the vessel wall associated with deposition in the immune complex, thus causing luminal narrowing or occlusion. It is mainly associated with class IV and primarily affects arterioles. The main characteristic is the deposition of fibrinoid material that expands the arterial intima and occludes the vascular lumen. Necrosis of myocytes and endothelium can occur; however, there is no inflammatory infiltrate. On immunofluorescence, IgG and fibrin deposits indicate the combination of immune complex deposits and intravascular coagulation. It presents clinically with severe hypertension and rapid loss of renal function, and it has a poor prognosis.
TMA. This is defined as vessel luminal narrowing caused by eosinophilic and fucsinophilic deposits positive for fibrins according to staining and is associated with endothelial oedema and luminal thrombi (acute TMA) or intimal mucoid oedema and onion skin intimal fibrodysplasia (chronic TMA). It affects small arteries, arterioles, and glomerular capillaries, and it may occur in different scenarios, such as antiphospholipid antibody syndrome, haemolytic uremic syndrome, thrombotic thrombocytopaenic purpura, scleroderma overlap syndrome, malignant hypertension, and the use of calcineurin inhibitors. It can also occur with any LN class or be an isolated finding (Figure 1 D).
True renal vasculitis. This is defined as fibrinoid necrosis of the arterial wall associated with infiltration of the vessel wall by inflammatory cells. It is rare in LN. It may occur in isolation or can be associated with systemic vasculitis and with any class of LN, regardless of glomerular activity.
Although the current ISN/RPS classification of LN does not evaluate vascular complications, a recent review suggested the importance of such an approach and of standardised terminology to distinguish arterial or common arteriolar sclerosis from lesions related to lupus, such as vasculopathy associated with the deposition of immune complexes, vasculitis, and TMA. Definitions of TMA and vasculitis in LN still need to be created because they may occur in an isolated manner with or without specific associated serological findings (antineutrophil cytoplasmic antibodies [ANCA), antiphospholipid antibodies, etc.), or they may coexist with glomerular injury mediated by imm(ne complexes. Therefore, it has been proposed that lupus vasculopathy should be defined as the luminal narrowing of arterioles or interlobular arteries through intramural immune deposits typically associated with fibrinoid changes without inflammation of the vessel wall and confirmed by immunofluorescence through the presence of immunoglobulins and complement fractions in deposits [4].
TMA is associated with LN in 0.5% to 10% of the cases. The incidence of LN class IV with TMA varies from 5% to 9% in the literature [27, 42]. In some studies, the presentation and evolution of LN in these patients are worse than those in patients with LN without TMA [43, 44]; however, this has not been demonstrated in other studies [30, 44, 45].
TMA may be the glomerular manifestation of antiphospholipid antibody syndrome (APS), which is an autoimmune disease characterised by the occurrence of venous and/or arterial thrombosis and gestational morbidity in the presence of pathogenic autoantibodies known as antiphospholipid antibodies (lupus anticoagulant and anticardiolipin antibodies) [46, 47]; APS is divided into primary and secondary. Furthermore, APS mainly occurs with cancer, infections, and autoimmune diseases, among which SLE stands out. The presence or absence of SLE may modify the clinical or serological expression of APS. In addition to the classic manifestations, patients with APS most commonly have arthralgia, arthritis, autoimmune haemolytic anaemia, livedo reticularis, epilepsy, myocardial infarction, and glomerular thrombosis [47].
APS nephropathy (APSN) is the result of thrombosis in renal arteries or veins, intraparenchymal arteries, and glomerular capillaries. Histologically, APSN is characterised by TMA, but chronic vaso-occlusive lesions are also commonly observed (fibrous intimal hyperplasia, focal cortical atrophy, fibrous occlusion of arteries) [48].
A 2002 study that retrospectively analysed renal biopsy samples from 114 patients with systemic lupus determined the following: APSN was present in 32% of renal biopsy samples in addition to and independently of the presence of NL, and it was statistically associated with the presence of lupus anticoagulant antibodies, but not with anticardiolipin antibodies; APSN is associated with extrarenal APS, mainly arterial thrombosis and obstetric foetal loss, but not with APS venous thrombosis; and APSN is an independent risk factor that contributes to a high prevalence of hypertension, elevated serum creatinine, and increased interstitial fibrosis. Therefore, it seems likely that APSN can worsen the prognosis of these patients [49].
In a recent retrospective analysis of 429 patients with lupus confirmed by biopsy, a total of 200 patients (46.6%) had no vascular complications according to the renal biopsy results, 89 (44.0%) had arterial sclerosis, 6 (1.4%) had non-inflammatory necrotising vasculopathy, 23 (5.4%) had TMA, and 11 (2.6%) had true renal vasculitis. Patients without renal vascular complications were younger and had better renal function. Patients with TMA and true renal vasculitis had worse renal function and higher blood pressure in the beginning of the study. The five-year kidney survival rates were 83% in the absence of renal vascular complications, 63% with arterial sclerosis, 67% with non-inflammatory necrotising vasculopathy, 31% for TMA, and 33% for true renal vasculitis. Non-inflammatory necrotising vasculopathy and true renal vasculitis were significantly correlated with activity scores, whereas chronic arterial sclerosis and TMA were correlated with chronicity scores. However, in this study, renal vascular complications were associated with worse renal outcomes but did not behave as independent factors [35].
Thus, it seems reasonable to include vascular lesions in the NIH activity and chronicity indexes. We suggest that TMA could be included in the activity index, while thickening of the intima of arterioles could be included in a semi-quantitative way in the chronicity index: < 25% of the lumen (1+), 25–50% of the lumen (2+), or > 50% of the lumen (3+), because this would add better prognostic value to these indexes. However, studies to validate this inclusion are still needed.
Lupus podocytopathy
Nephrotic syndrome is a common sign of LN, which is usually associated with the deposition of immune complexes in the glomerular capillary wall. It is often accompanied by endocapillary proliferation or necrosis and is particularly characteristic of proliferative LN (classes III and IV) or membranous LN (class V) [50, 51]. However, it was observed some years ago that a subset of SLE patients who presented with nephrotic syndrome were found to have normal glomeruli or only mild glomerular mesangial. For these patients, the most prominent electron microscopy finding was diffuse effacement of the podocyte processes without immune deposits in the peripheral capillary wall, suggesting podocytopathy. With the exception of immune deposits in the mesangial region, glomerular podocytopathy in these patients shared similar characteristics with minimal change disease (MCD), which was previously classified as SLE with idiopathic MCD [52, 53]. Accumulated evidence from clinical and epidemiological studies suggested that MCD in these patients was indeed related to SLE rather than being concomitant with SLE [54–56].
Thus, lupus podocytopathy is currently defined as SLE patients clinically presenting with nephrotic syndrome, demonstrating minimal change disease (MLD), mesangial proliferation (MsP) or focal segmental glomerulosclerosis (FSGS) on optic microscopy, while diffuse podocyte foot process effacement in the absence of subepithelial or subendothelial deposition is the only morphological feature on electronic microscopy [57]. However, the commonly used International Society of Nephrology/Renal Pathology Society classification of LN does not include lupus podocytopathy [1].
Hu et al. studied of 3750 biopsy samples from patients with LN in China [58] and identified 50 cases (1.33%) of lupus podocytopathy, including 13 cases of MCD, 28 cases with evidence of mesangial proliferation, and 9 cases of focal segmental glomerulosclerosis (FSGS). Diffuse podocyte process effacement was evident in all 50 biopsy specimens, and electron dense deposits present exclusively in mesangial cells were evident in 47 biopsy specimens. All 50 patients had nephrotic syndrome and 34% had acute kidney injury (AKI). Most notably, patients with FSGS were at higher risk for AKI and severe tubulointerstitial injury and had lower rates of remission than patients with MLD/mesangial proliferation, although no patient died or developed CKD over a mean follow-up period of 62 months. Based on differences in the incidence of AKI, severity of tubular injury, and treatment response, the researchers proposed that lupus podocytopathy be divided into two different subtypes: MLD/mesangial proliferation and FSGS.
For this reason, Hu et al. [58] proposed criteria for diagnosing lupus podocytopathy based on clinical presentation and hallmark biopsy findings on light microscopy, immunofluorescence microscopy, and electron microscopy. Lately, Bomback and Markowitz [59], based on a paper by Hu et al., described fairly simple criteria to diagnose lupus podocytopathy: (1) clinical presentation of full nephrotic syndrome in a patient with SLE, (2) diffuse and severe foot process effacement, and (3) the absence of subendothelial or subepithelial immune deposits.
Their purpose was to separate the classic forms of immune complex-mediated LN from lupus podocytopathy, with a willingness to diagnose both in the appropriate situation [58, 59]. Thus, mesangial deposits and mesangial proliferation were excluded of the criteria and, if these findings are present, then the additional diagnosis of mesangial proliferative LN (LN class 2) is merited. If mesangial deposits are not accompanied by mesangial proliferation, the diagnosis of minimal mesangial LN (LN class 1) is rendered. They also subdivide lupus podocytopathy into patients who would otherwise meet criteria for MCD or FSGS, including the morphologic subtypes of FSGS (collapsing, tip lesion, etc.).
Also worthy of attention are the severe cases of nephrotic syndrome with rapid loss of renal function, usually associated with collapsing forms of FSGS (Figure 1 E), also known as collapsing glomerulosclerosis (CG). CG has a pattern of glomerular lesions and is characterised by severe podocytes injury, loss of cell differentiation markers, proliferation of podocyte cells and/or parietal epithelial cells filling the space of Bowman, and global or segmental collapse of the capillary tufts. These lesions are particularly unresponsive to standard therapies. Described for the first time as an idiopathic disorder or post-HIV infection, CG is now associated with a large group of diseases and different pathogenic mechanisms involved in podocyte injury and mitogenic stimulation that occur in LN [60–62].
In 2012, Salvatore et al. [63] reported a retrospective series of 19 LN patients with negative serology for HIV, hepatitis C, and parvovirus, who underwent renal biopsy due to proteinuria and, often, renal insufficiency, which showed CG with or without concomitant LN. These findings raised important questions about the pathogenesis of CG in this clinical scenario, and about the relationship of these lesions with lupus podocytopathy. CG that occurs in the scenario of LN shares certain demographic, clinical, morphological, and immunohistological characteristics with idiopathic CG and HIV: strong association with people of African descent; typical clinical presentation includes significant proteinuria and, usually, renal insufficiency; frequent progression to chronic kidney disease, especially in the absence of remission induced by antiproteinuric therapy; frequent tubular lesions and tubulointerstitial fibrosis in addition to glomerular injury characteristic of CG; loss of certain podocyte differentiation markers, especially in clusters with CG injury; and proliferation of glomerular epithelial cells, as indicated by Ki67 staining. However, CG also has different characteristics, especially at the molecular level, suggesting unique pathogenic aspects of CG in LN.
The pathogenesis of the remaining cases of CG in LN described by Salvatore et al. and other authors [55–57, 63–67] is not yet clear; however, it should be noted that the majority of these patients presented active systemic disease, and that a significant number had LN concomitantly, although it was usually mild (mostly LN class II according to ISN/RPS 2003). This certainly suggests the involvement of humoral and/or cell-mediated immunity, both of which are implicated in the pathogenesis of podocytopathies and SLE. The relationship between lupus podocytopathy and CG lesions remains unclear, as is the case for collapsing and non-collapsing forms of idiopathic FSGS, although for all these lesions the primary target is probably the podocyte; in fact, CG may represent an extreme form of lupus podocytopathy [68].
We consider the report from Hu et al. [58] and Bomback and Markowitz [59] to be important contributions that bring us closer to recognising and understanding the entity of lupus podocytopathy. The process of developing consensus diagnostic criteria should be straightforward, and a goal for the future will be to connect MCD and FSGS with SLE in more than an observational manner, as it has been done so far. For now, we have reached the point that lupus podocytopathy should be considered a distinct and recognisable disease entity, whose recognition should be added to the next revision of the classification of LN.
Crescents
The term “crescent” is used for a lesion consisting of extracapillary hypercellularity, composed of a variable mixture of cells predominantly born from epithelial proliferation or predominantly of monocytes and/or macrophages (Figure 1 F). Fibrin and fibrous matrix may also be present [4].
Crescent formation is a commonly observed pathological finding in LN. Literature on the prognostic significance of crescents formation in LN is limited, and only a handful of studies have assessed the unfavourable outcomes of crescentic nephritis wherein the crescents occupy > 50% of the glomeruli [69–71]. Therefore, current views on the prognosis of crescentic LN are controversial.
A retrospective cohort study compared crescentic LN with non-crescentic LN (class IV–G) and revealed that patients with crescentic GN had higher prevalence of acute kidney injury, higher pathological activity and chronicity scores, and higher relapse rates. Over a mean follow-up of 4 years, patients with crescentic LN had a significantly poorer renal outcome compared to those with non-crescentic class IV LN (incidence of renal-endpoint events: 48.5% vs. 11.0%, p < 0.001), with no obvious difference in long-term patient survival [69]. Conversely, Tang et al. [71] followed up 79 crescentic LN patients for 56.1 ±18.8 months and found that patients with diffuse crescent formation had similar clinical outcomes and renal survival to those with more severe LN (class IV), with 88.3% and 81.5% of patients preserving stable renal function at 5 and 8 years, respectively.
Given the heterogeneity of the disease itself, the investigated populations, treatment regimens, responsiveness to therapy, and follow-up duration, it is not surprising that the outcomes of crescentic LN patients have been incompatible. More recently, Zhang et al. [72] assessed the long-term prognosis of LN patients with different proportions of crescents. They showed that the LN patients with crescents were associated with much more severe baseline clinical status than those of LN patients without crescents. However, the long-term renal and patient survival appeared to be comparable, probably due to the fact that crescentic lesions usually occurred in class IV LN, implying that the intensive immunosuppression therapy might account for the overall comparable prognosis. Nevertheless, they observed that for LN patients with crescents, each increasing 10% proportion of crescents carried a higher risk for adverse outcomes of renal progression and mortality, and crescentic LN exhibited the worst patient survival.
The definition of extracapillary proliferation or a cellular crescent given in the ISN/RPS classification is “extracapillary cell proliferation of more than two cell layers occupying one fourth or more of the glomerular capsular circumference”. This definition only holds for a cellular crescent; fibrocellular and fibrous crescents lack a definition. Fibrocellular and fibrous crescents are only mentioned in Table 6 in the ISN/RPS classification paper, which states that both cellular and fibrocellular crescents are regarded as active lesions and that fibrous crescents are regarded as chronic lesions [1], and how extracapillary proliferation contributes to determining whether a biopsy falls into either the IV-S or IV-G subcategory is a complicated issue. Regarding this matter, Wilhelmus et al. [3] defined that extracapillary proliferation can never contribute to the segmental or global nature of a class IV lesion. If we want to include extracapillary proliferation when assessing the segmental or global nature of the lesion, the area should be redefined in which both endocapillary and extracapillary lesions can occur, to establish whether we are dealing with segmentally or globally affected glomeruli. Finally, the term extracapillary proliferation holds some of the same objections as the term endocapillary proliferation. Therefore, one could consider using the term extracapillary hypercellularity rather than extracapillary proliferation.
The ISN/RPS criterion of a crescent involving 25% or more of the glomerular capsular circumference also was discussed by Bajema et al. in 2018 [4]. It was decided that this threshold should be 10% or more in accord with evidence from the Oxford Classification of IgA nephropathy and standard approaches used in clinical reporting of renal biopsy lesions. Crescents should be composed of more than two cell layers in order to distinguish them from apposition of the single layers of hypertrophied visceral and parietal cells. The group proposed definitions for the distinction of cellular, fibrous, and fibrocellular crescents, which were lacking in the ISN/RPS lupus nephritis classification, and also defined that extracapillary hypercellularity attributable to concurrent collapsing glomerulopathy lesions should not be designated as crescents [4].
Overlap syndromes
Although overlap syndromes are not necessarily linked to additional histopathological lesions in LN, their presence carries additional prognostic value to the patient with lupus. Therefore, their aspects will be summarised below.
SLE and related ANCA vasculitis
Although ANCA were initially associated with primary vasculitis, approximately 15% to 20% of patients with lupus have detectable ANCA. ANCA are most commonly found in SLE patients and are even more common in LN patients than in patients with other clinical manifestations of SLE, and, furthermore, they have been associated with diffuse proliferative LN, particularly class IV-S rather than class IV-G [73–75].
Pauci-immune glomerulonephritis in SLE was first described in 1983 in 4 patients with minimal serologic evidence of disease activity [76]. Since then, the relevance of ANCA in the pathophysiology of SLE has been controversial and the association between ANCA positivity and LN activity, histological features, or prognosis remains debatable. Clinicians and renal pathologists often do not know how to interpret the discovery of clinically and serologically confirmed SLE and ANCA positivity [77].
ANCA seem to influence the histological pattern of LN and are associated with worse kidney function at baseline and more active lupus serology. In fact, a body of literature suggests that in patients with lupus nephritis, ANCA positivity may correlate with a specific clinical and histological subset: necrotising and crescentic glomerulonephritis and more severe renal disease [78–81].
Turner-Stokes et al. [79] retrospectively studied standardised reports of renal biopsies, which, combined with clinical data, support the notion that the positivity of ANCA in patients with LN is associated with an alleged phenotype of more active lupus diseases, as indicated by higher levels of antibodies to double-strand DNA, as well as more severe glomerular lesions, including glomerular crescents, reflecting worse activity index.
However, the majority of studies that correlated SLE to ANCA positivity are retrospective and have several limitations. Therefore, further studies are needed to examine whether ANCA positivity in patients with LN has a pathogenic role and is associated with the worst renal outcomes or is simply a marker of more severe condition, especially when comparing patients undergoing similar treatment modalities.
SLE and scleroderma
Overlap of systemic sclerosis (SSc) with SLE is a frequent presentation in clinical practice, although their co-occurrence does not get the attention it deserves in the literature, even when it is related in 1–27% of cases, depending on the study cohort [82–84].
The real challenge for diagnosis and treatment of SSc and SLE overlap syndrome arises among patients with renal involvement, with signs and symptoms that can be manifestations of lupus nephritis or, more rarely but just as importantly, a complication of SSc, such as scleroderma renal crisis (SRC), because both can progress with rapid loss of kidney function [85]. New-onset accelerated hypertension with progressive oliguric renal failure is the usual presentation of SRC [86], which is the result of endothelial cell damage and vasospasm culminating in fibrinoid necrosis of kidneys, featuring TMA. This causes activation of the renin-angiotensin-aldosterone system and hypertension in these patients [87] and could progress to deterioration of kidney function with non-nephrotic proteinuria and haematuria, microangiopathic haemolytic anaemia, and thrombocytopaenia [88].
On the other hand, glomerular involvement with immune complex deposit, proteinuria, and haematuria with decline in kidney function, as well as concomitant hypertension and haemolytic anaemia are well-known findings in SLE [89]. The presence of rich urinary sediments with low complement level, Coombs test positivity, and absence of schistocytes in contrast to microangiopathic haemolytic anaemia can help in the differential diagnosis of SLE from SRC. In the meantime, laboratory evaluations cannot replace information that can be obtained only from renal biopsy in a patient with overlap serology, because serological markers are not always helpful in differential diagnosis [90].
Conclusions
Although the ISN/RPS 2003 classification of LN has gained international recognition, the reduced exploration of other mechanisms of renal injury in the pathogenesis of renal involvement by SLE has been the target of criticism. It is known that the presence of specific lesions may have therapeutic and prognostic implications. For example, interstitial inflammation is potentially reversible; the presence of interstitial nephritis is an important histological finding that may identify patients at high risk for progression to chronic kidney disease; the presence of lupus podocytopathy, especially in the form of collapsing glomerulosclerosis, may represent an extreme form of podocyte damage, indicating a worse prognosis; and data regarding the prognostic value of renal vascular complications are conflicting, thus necessitating increasingly concise descriptions and grading of these lesions. Therefore, it is suggested that renal involvement by SLE, in addition to lupus glomerulonephritis, should be studied further and that new knowledge regarding the pathogenesis of the disease should be incorporated into the current classification to improve the clinical value and help with therapeutic decision-making, thus allowing better outcomes for patients with LN.