Introduction
Mysterious pneumonia has been reported during December 2019. The causative agent for this pneumonia was identified to be a novel coronavirus (nCoV) named earlier as 2019-nCoV and now termed coronavirs 2). The disease caused by SARS-CoV-2 was named as coronavirus disease 2019 (COVID-19). This virus belongs to the subfamily Coronavirinae; this group of viruses is known to cause respiratory and gastrointestinal infections [1]. The first coronavirus was reported in the 1960s because it caused mild respiratory symptoms; these viruses were given the names (i) human CoV 229E (HCoV-229E) and (ii) HCoV-OC43 [2]. In 2003, sever acute respiratory syndrome-Cov (SARS-CoV) was identified as the agent that caused a global pandemic [3]. As of 03:00 GMT+3, 9 April 2020, more than 200 countries have been affected around the world, with a total of 1,436,198 diagnosed cases and 85,522 deaths [4]. Compared to the coronavirus reported to cause an outbreak in the Middle East, so-called MERS-CoV, the mortality rate for SARS-CoV-2 is relatively low; however, the number of affected cases is extremely high [2, 4, 5]. The latest SARS-CoV-2 was isolated by Chinese scientists, and the genetic sequence of the virus is now available [6]. The new outbreak is believed to be related to meat that derived from wild or captive sources at a seafood market [7]. All of the human CoV are known to have an animal source: SARS-CoV, MERS-CoV, HCoV-NL63, and HCoV-229E are considered to have originated from bats; HCoV-OC43 and HKU1 probably originated from rodents [8] The recurrent corona outbreaks pose a global health threat as a new outbreak could not be avoided due to the interaction between human and animals, as well as human to human contact [1]. This urges the need to develop new curative treatment strategies and possible vaccines.
Helicase is a viral replication enzyme in coronavirus, which has the characteristics of unwinding DNA and RNA and separating them into two single-stranded nucleic acids [9, 10]. These vital characteristics of coronavirus helicase describe the importance of helicase as a druggable target [9–11]. In the present study, SARS-CoV-2 helicase was selected to identify helicase inhibitors by using computational state-of-the-art tools to identify drug targets among clinically approved drugs. The study suggests the best active clinically approved drugs against SARS-CoV-2 helicase through molecular docking analysis.
Material and methods
Amino acid sequence retrieval and prediction of secondary structure
The ORF1ab polyprotein sequence of SARS-CoV-2 isolate 2019-nCoV/USA-WA1-F6/2020 (GenBank: QHU79203.1) was retrieved from NCBI database on 12 Feb 2020 (Figure 1). The helicase region was selected based on the amino acid sequence alignment using MAFFT [12] and MEGA7 [13] with Bat SARS, SARS-CoV, MERS-CoV, and other sequences. Phylogeny was constructed (Figures 2 and 3). For the homology modelling, the sequences of helicase were searched against the protein database using BLAST-P [14]. The proteins having PDB Id: 6jyt.2.A [15] and 5wwp.1.A [16] were selected for use as templates for 3D modelling of helicase protein of SARS-CoV-2.
Figure 1
Flow chart of the methodology for the selection of the best inhibitor of SARS-CoV-2 helicase
Figure 2
Phylogenetic analysis of SARS-CoV-2 helicase protein using RefSeq-protein BLAST results by maximum likelihood method. “The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model [33]. The bootstrap consensus tree inferred from 500 replicates [34] is taken to represent the evolutionary history of the taxa analysed [34]. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. Initial tree(s) for the heuristic search was/were obtained automatically by applying Neighbour-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The analysis involved 101 amino acid sequences. All positions containing gaps and missing data were eliminated. There were a total of 74 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 [13].” Sequence of COVID-19 helicase used for the phylogenetic analysis: DAVVYRGTTTYKLNVGDYFVLTSHTVMPLSAPTLVPQEHYVRITGLYPTLNISDEFSSNVANYQKVGMQKYSTLQGPPGTGKSHFAIGLALYYPSARIVYTACSHAAVDALCEKALKYLPIDKCSR IPARARVECFDKFKVNSTLEQYVFCTVNALPETTADIVVFDEISMATNYDLSVVNARLRAKHYVYIGDPAQLPAPRTLLTKGTLEPEYFNSVCRLMKTIGPDMFLGTCRRCPAEIVDTVSALVYDN LKAHKDKSAQCFKMFYKGVITHDVSSAINRPQIGVVREFLTRNPAWRKAVFISPYNSQNAVASKILGLPTQTVDSSQGSEYDYVIFTQTTETAHSCNVNRFNVAITRAKVGILCIMSDRDLYDKL FTSLEIPRRNVATLQAENVTGLFKDCSKVITGLHPTQAPT
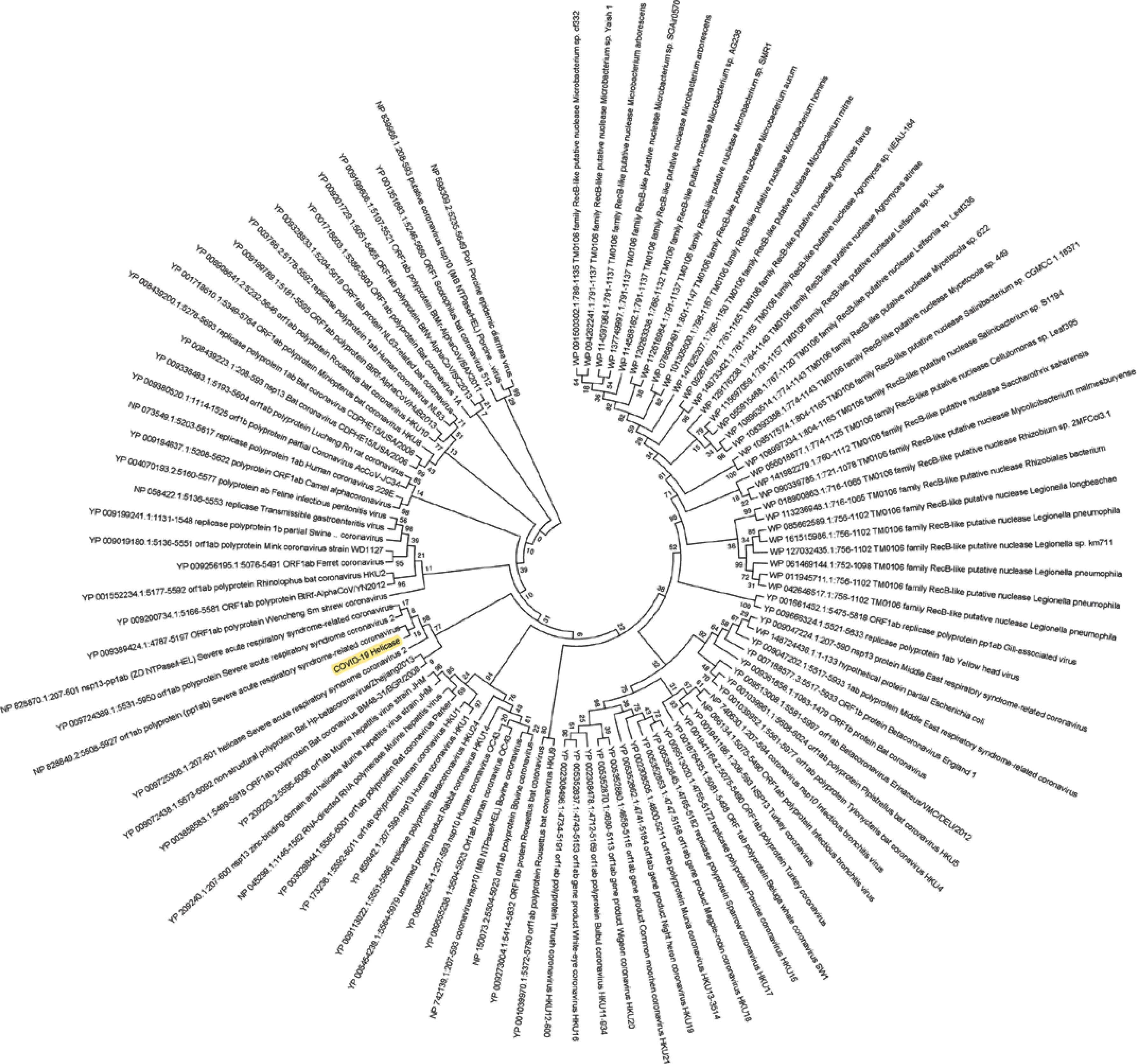
Figure 3
Phylogenetic analysis by Maximum Likelihood method of SARS-CoV-2 helicase protein using PSI-BLAST results. “The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix- based model [34]. The tree with the highest log likelihood (–3764.53) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbour-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The analysis involved 501 amino acid sequences. All positions containing gaps and missing data were eliminated. There were a total of 384 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 [13]”
Prediction and validation of 3D structure
An automated SWISS MODEL server was used to predict the structure by homology modelling [8]. The modelled PDB was visualised using PYMOL [17] and validated using PROCHECK [18]. Ramachandran plot statistics using the PDBsum structural analysis server was used to validate the 3D models [18]. The best model among the generated models was selected based on the criteria of having the minimum number of residues in the disallowed region and the highest number of residues in the allowed region, which was considered as a suitable model for helicase protein of SARS-CoV-2, and the same was used for further analysis. In the validated model, 3D atomic coordinates of the receptor were used to verify potential sites for binding of clinically approved drugs and docking.
Clinically approved drugs and molecular docking
Structure data of all the clinically approved drugs (such as protease inhibitors, nucleoside reverse transcriptase inhibitors, and non-nucleoside reverse transcriptase inhibitors) used to treat HIV (human immunodeficiency virus) infection were taken from the DrugBank and PubChem databases (Table I) [19, 20]. All the clinically approved drugs screened in the study are listed in Table I with the mechanism of action based on DrugBank and others [19–21 http://www.myhealth.gov.my/en/hiv-theraphy/]. The clinically approved drugs were obtained from the PubChem database as structure-data file (SDF) format, which were used in MOE software. Energy minimisation was completed through MOE tools followed by protonation the structure with default parameters [21, 22]. The selected approved drug molecules were subjected to a Lipinski filter and used for further analysis. Helicase-approved drug interaction analysis was done through flexible molecular docking by 3D protonation followed by energy minimisation in MOE software [23].
Table I
The molecular docking of approved drugs on the market against helicase of SARS-CoV-2
| Approved drug | Mechanism of action as per DrugBank | ΔG [kcal/mol] | Log P | NHB | HbA | HbD | Binding or interactive res-idues helicase | Strain Solvation Affinity | S | rmsd_refine | E_conf | E_place | E_score1 | E_refine | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vapreotide** | Analgesic, treatment of AIDS-related diarrhoea | –11.58 | 2.01 | 1 | 1 | 0 | GLY79 | 28.64 | –89.67 | –12.88** | –9.84** | 2.44 | 26.14 | 33.62 | –2.93 | –59.79 |
| Atazanavir* | Protease inhibitor, treatment of HIV infection | –6.6 | 4.48 | 1 | 1 | 0 | GLN331 | 15.78 | –75.03 | –11.28* | –9.32* | 2.43 | 4.91 | –26.22 | –7.83 | –50.84 |
| Lopinavir | Protease inhibitor, treatment of HIV infection | –6.75 | 4.33 | 3 | 2 | 1 | GLU331, ALA107 | 12.67 | –68.46 | –10.71 | –8.48 | 1.55 | –58.70 | –42.48 | –7.96 | –45.21 |
| Nelfinavir | Protease inhibitor, treatment of HIV infection | –7.39 | 4.75 | 1 | 0 | 1 | LYS82 | 11.66 | –69.05 | –10.7 | –7.68 | 2.12 | 22.20 | –46.40 | –8.34 | –38.47 |
| Saquinavir | Protease inhibitor, treatment of HIV infection | –7.68 | 3.09 | 1 | 1 | 0 | ALA107 | 37.65 | –72.56 | –10.6 | –7.79 | 1.57 | 0.76 | –81.02 | –8.77 | –36.78 |
| Colistin | Polymyxin antibiotic agent | –7.46 | –5.83 | 2 | 1 | 1 | GLU334, ARG237 | 26.67 | –70.15 | –10.31 | –9.09 | 4.10 | 53.75 | 58.81 | 2.70 | –54.68 |
| Indinavir | Protease inhibitor, treatment of HIV infection | –4.71 | 3.23 | 1 | 1 | 0 | ARG237, | 19.63 | –64.4 | –10.04 | –8.29 | 2.82 | 59.33 | –66.90 | –7.08 | –44.09 |
| Glecaprevir | Protease inhibitor, Treatment of HCV | –7.43 | 4.17 | 3 | 3 | 0 | ARG237, ARG236 | 20.49 | –63.01 | –10 | –7.50 | 1.36 | 230.70 | –56.50 | –7.21 | –36.20 |
| Ritonavir | Protease inhibitor, treatment of HIV infection | –6.29 | 6.44 | 1 | 0 | 1 | ALA110 | 13.55 | –59.7 | –9.39 | –7.90 | 2.06 | –97.39 | –63.16 | –7.95 | –45.40 |
| Aprepitant | Inhibitor of the HCV NS3/4A protease | –6.83 | 5.78 | 2 | 2 | 0 | ARG237 | 19.72 | –61.54 | –8.61 | –6.92 | 2.31 | 143.88 | –74.13 | –8.92 | –41.91 |
| Perphenazine | Antipsychotic drug | –4.42 | 3.94 | 3 | 2 | 1 | ARG237, GLU169 | 3.88 | –57.34 | –8.61 | –7.04 | 1.66 | 113.07 | –62.40 | –10.83 | –35.37 |
| Darunavir | Protease inhibitor, treatment of HIV infection | –4.37 | 2.38 | 1 | 1 | 0 | ARG237 | 8.93 | –52.82 | –8.52 | –7.52 | 1.67 | –19.73 | –56.20 | –9.29 | –42.46 |
| Rilpivirine | NRTI, treatment of HIV infection | –5.97 | 4.99 | 2 | 2 | 0 | ARG236 | 5.11 | –51.79 | –8.03 | –7.25 | 2.10 | –139.36 | –82.74 | –9.96 | –43.80 |
| Delavirdine | NRTI, treatment of HIV infection | –3.08 | 2.72 | 3 | 3 | 0 | ARG236, 236, 236 | 14.17 | –49.59 | –7.96 | –7.35 | 1.57 | –25.78 | –64.65 | –11.89 | –41.74 |
| Abacavir | NRTI, treatment of HIV infection | –2.76 | 1.19 | 2 | 0 | 2 | GLN331, ALA 107 | 7.08 | –47.06 | –7.93 | –6.52 | 1.40 | 31.33 | –68.30 | –9.12 | –27.75 |
| Hydroxychloroquine# | Currently used | 3.78 | –3.26 | 1 | 1 | 0 | GLY332 | 3.24 | –46.05 | –7.71 | –6.33 | 2.26 | –19.61 | –49.01 | –10.09 | –29.27 |
| Didanosine | NRTI, treatment of HIV infection | –1.42 | 0.24 | 1 | 0 | 1 | GLN331 | 14.13 | –46.22 | –7.21 | –5.73 | 1.72 | 78.33 | –55.16 | –8.42 | –30.16 |
| Zidovudine | NRTI, treatment of HIV infection | –0.66 | 0.23 | 2 | 2 | 0 | LYS82, ALA107 | 11 | –68.28 | –7.16 | –6.21 | 1.65 | –117.16 | –66.87 | –10.27 | –27.68 |
| Tenofovir | NRTI, treatment of HIV infection; effective against herpes simplex virus 2, and HBV | –0.96 | –0.86 | 3 | 2 | 1 | SER83, GLY79, ARG237 | 12.73 | –47.51 | –7.1 | –5.85 | 1.36 | –59.95 | –54.86 | –10.39 | –29.40 |
| Etravirine | NRTI, treatment of HIV infection | –6.75 | 4.72 | 3 | 3 | 0 | ARG236, ARG237, GLY79 | 5.97 | –44.53 | –6.98 | –6.57 | 2.88 | –103.48 | –41.18 | –11.50 | –38.76 |
| Stavudine | NRTI, treatment of HIV infection | –0.8 | –0.28 | 4 | 2 | 2 | ASP168, LYS82, GLY332 | 6.55 | –36.56 | –6.4 | –5.48 | 1.25 | –105.35 | –51.51 | –9.20 | –18.96 |
| Zalcitabine | NRTI, treatment of HIV infection | –0.88 | –0.21 | 1 | 1 | 0 | GLY79 | 4.72 | –38.13 | –6.34 | –5.66 | 1.06 | –126.84 | –68.31 | –9.93 | –26.98 |
| Nevirapine | NRTI, treatment of HIV infection | –2.28 | 2.65 | 1 | 1 | 0 | ARG237 | 2.19 | –34.28 | –6.27 | –5.38 | 1.73 | 79.43 | –63.26 | –8.11 | –23.68 |
| Emtricitabine | NRTI, treatment of HIV infection | –1.74 | 0 | 2 | 2 | 0 | ARG237, ARG237 | 14.78 | –38.95 | –6.02 | –5.08 | 2.01 | –47.31 | –53.29 | –8.40 | –23.52 |
| Lamivudine | NRTI, treatment of HIV infection | –1.35 | –0.3 | 1 | 1 | 0 | ARG236 | 4.76 | –34.4 | –5.96 | –4.95 | 1.67 | –117.15 | –78.49 | –8.85 | –23.27 |
| Favipiravir# | Currently approved anti–influenza drugs | –1.33 | –1.19 | 1 | 1 | 0 | ARG267 | 3.66 | –23.99 | –4.65 | –4.47 | 1.76 | 41.22 | –58.42 | –7.91 | –17.86 |
The most significant approved drug with lowest binding affinity, lowest MOE Score (S: the final score of GBVI/WSA binding free energy) (kcal/ mol) and obeying Lipinski’s rule (NHB – number of hydrogen bonds; HbA – hydrogen bond acceptor; HbD – hydrogen bond donors; Log P: –The log octanol/water partition coefficient); AIDS – acquired immunodeficiency syndrome; HIV – human immunodeficiency virus; NRTI – nucleoside reverse transcriptase inhibitor; HCV – hepatitis C virus; HBV – hepatitis B virus; NNRTI – non-nucleoside reverse transcriptase inhibitors. RMSD_refine – the mean square deviation after refinement; E_place – score of the placement phase; E_conf – energy conformer; E_refine – score refinement; E_scor1 – score the first step of notation.
* drug with the second lowest binding energy and S score; #currently used antiinfluenza drug. Active sites of SARS-CoV-2 helicase are GLU55 PHE56 SER58 ASN59 PRO77 PRO78 GLY79 THR80 GLY81 LYS82 SER83 HIS84 ALA107 ALA110 LEU111 LYS114 ASP168 GLU169 GLN198 LEU232 GLY233 THR234 ARG236 ARG237 GLN331 GLY332 SER333 GLU334 ARG361.
Results
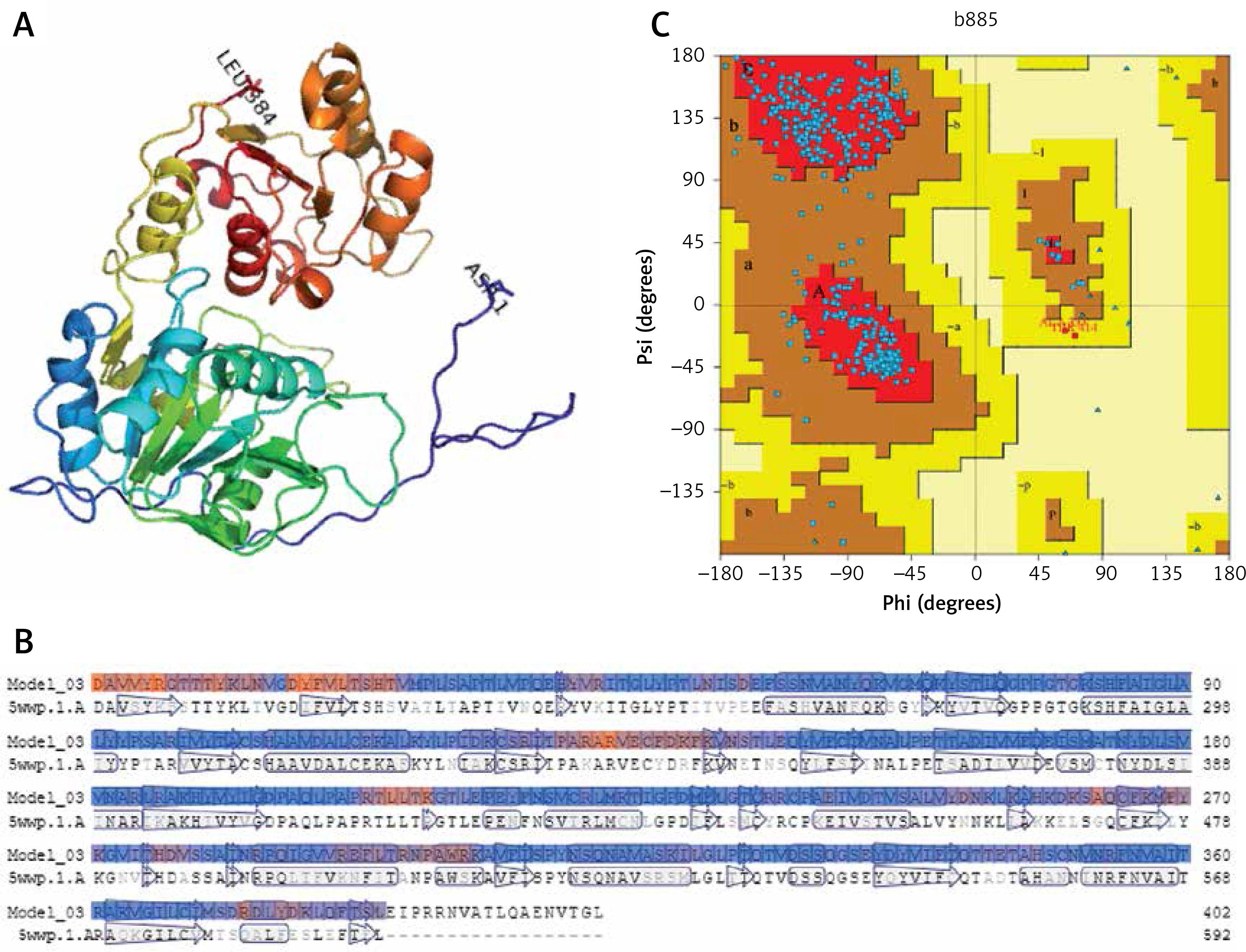
The amino acid sequences of 2019-nCoV/USA-WA1-F6/2020 (GenBank: QHU79203.1) helicase region were blasted against the PDB-BLAST database to identify a proper template for homology modelling. Helicase has been considered as a target by similarity screening through phylogenetic analysis of SARS-CoV-2 protein sequence (420 amino acids) from the data retrieved through RefSeq protein BLAST and PSI-BLAST, which revealed that the helicase sequence is similar to helicase protein of severe acute respiratory syndrome-related coronavirus and bat coronavirus (Figures 2 and 3). The proteins having PDB ID: 6jyt.2.A and 5wwp.1.A were checked initially as appropriate templates for 3D modelling of helicase protein region in SARS-CoV-2, and finally 6jyt.2.A was selected as an appropriate template due to 99.78% sequence identity between 6jyt.2.A and the query. The 3D structure of helicase protein was built based on the target-template alignment using ProMod3 by SWISS MODEL. The best model of helicase protein was selected with QMEAN-score –1.71 and highest resolution 3.00Å, and the model was validated using PDBsum structural analysis. The helicase protein modelled structure’s stereochemical stability was estimated through a Ramachandran plot, which indicated that the 3D structure of SARS-CoV-2 helicase protein is a good quality model with 89.4% amino acid residues of predicted helicase structure in the favoured region and the remaining amino acid residues in the allowed region (10.6%); furthermore, none of the residues are in the disallowed regions (Figure 4).
Figure 4
Homology model of helicase. A – Helicase homology model. B – Model-Template Alignment. C – Ramachandran plot of SARS-CoV-2 helicase from PDBsum
Molecular docking
The modelled helicase protein of SARS-CoV-2 was analysed by MOE software for the helicase-approved drug interaction, initially by 3D protonation, energy minimisation and prediction of active site and properties for the 21 clinically approved drugs using default parameters. The SARS-CoV-2 helicase protein docked with the 21 anti-HIV clinically approved drugs. Furthermore, the recently approved anti-influenza drug (favipiravir) and widely used hydroxychloroquine were docked separately in MOE (Figure 5, Table I).
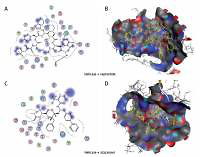
Figure 5
The most significant drug – SARS-CoV-2 helicase interaction. A, B – SARS-CoV-2 helicase and vapreotide interaction; A – 2D plot of SARS-CoV-2 helicase and vapreotide interaction; B – 3D structure of saquinavir – SARSCoV- 2 helicase interaction. C – 2D plot of SARS-CoV-2 helicase and atazanavir interaction. D – 3D plot of SARS-CoV-2 helicase and atazanavir interaction. E – Binding affinity and MOE score (S) of available drugs and interaction with SARS-CoV-2 helicase. **Drug with the lowest binding affinity and S score or binding free energy; *drug with the second lowest binding affinity and S score or binding free energy. Details of the interaction can be seen in Table I
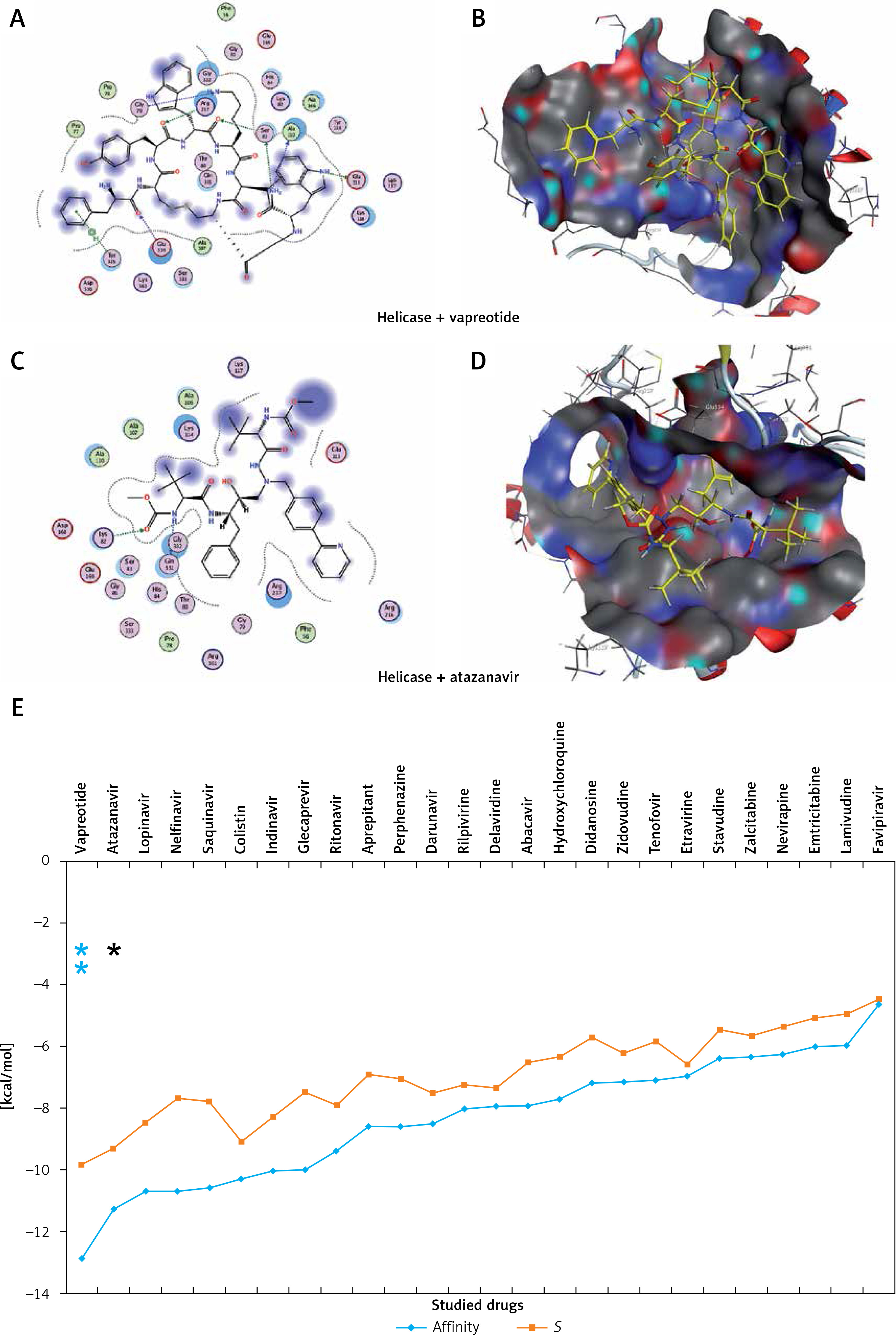
The 23 input clinically approved drugs were screened based on binding affinity, London dG scoring function and MOE score or GBVI/WSA binding free energy (S, kcal/mol). Table I is sorted according to binding affinity and MOE score (S, binding free energy kcal/mol). The helicase-approved drug interaction through molecular docking predicted that vapreotide (affinity: –12.88; S score: –9.84 kcal/mol) and atazanavir (affinity: –11.28; S score: –9.32 kcal/mol) are the most potent inhibitors of helicase of SARS-CoV-2 amongst clinically approved drugs (Table I). Furthermore, we visualised the interaction between vapreotide and atazanavir and SARS-CoV-2 helicase, as shown in Figure 5. The residue position of GLY79 and GLN331 of SARS-CoV-2 helicase protein showed the hydrogen bond with vapreotide and atazanavir, respectively (Figures 5 A, C) and it shows a strong affinity (Table 1). The active residue of helicase, GLY79, and GLN331 were satisfactory at a distance of 2.44Å and 2.43 Å from bound vapreotide and atazanavir, respectively (Table I). Furthermore, both predicted drugs satisfy the condition of the Lipinski rule of five, such as partition coefficient (logP), hydrogen bond acceptor, and donor (Table I).
Discussion
Coronavirus disease 2019 (COVID-19) cased by SARS-CoV-2 (previously 2019-nCoV) is a global pandemic health threat [4, 7, 8, 24–26]. The present rapid molecular docking study was carried out considering the extreme health and economic problems arising due to COVID-19 and the consequent high mortality all over the world, to screen anti-SARS-CoV-2 drugs among approved drugs for treating HIV infection. Characterisation and biochemical properties of helicase in severe acute respiratory syndrome CoV showed that it unwound DNA and RNA [9, 11]. Helicase enzyme in coronavirus is a prominent viral replication enzyme. Helicases are evolutionarily conserved proteins in coronaviruses and Nidovirales [27]. Furthermore, double-stranded nucleic acids are separated into two single-stranded nucleic acids by helicases, which catalyse the separation [10]. Earlier studies have described the importance of coronavirus helicase on the therapeutic target [10]. Helicase can hydrolyse all ribonucleotide triphosphates and deoxyribonucleotide triphosphates in the SARS coronavirus [9, 11]. Helicase enzyme in coronavirus increases the unwinding of nucleic acid by twofold [10]. Hence, SARS-CoV-2 helicase was selected to identify helicase inhibitors through state-of-the-art tool-based screening to reveal the anti-SARS-CoV-2 drug targets. We used 23 clinically approved drugs previously listed for the treatment of HIV infection [19, 21]. The phylogenetic analysis of SARS-CoV-2 helicase amino acid (420 amino acid) sequence against various sequence data retrieved through RefSeq protein BLAST and PSI-BLAST revealed > 90% similarity with SARS CoV in molecular phylogenetic analysis by maximum likelihood method with 500 replications in bootstrap [13]. Furthermore, the selected (PDB Id: 6jyt.2.A) template from severe acute respiratory syndrome coronavirus [15] showed 99.78% sequence identity with the SARS-CoV-2 helicase.
Currently used drugs [28], favipiravir-SARS-CoV-2 helicase interaction, and hydroxychloroquine-SARS-CoV-2 helicase interaction have less binding affinity compared to most of the drugs screened in the study. The SARS-CoV-2 helicase and approved drug interaction using the modelled and validated druggable helicase protein predicted vapreotide and atazanavir as targets among the 23 approved drugs, as medications for HIV infection. Atazanavir is a protease inhibitor that is used to treat HIV. It inhibits HIV-1 protease, which is needed to cleave the viral polyprotein precursors. The absence of cleavage results in immature viral particles [29, 30]. Vapreotide was used for treating patients with AIDS-related diarrhoea [31], which showed the lowest binding free energy interaction with SARS-CoV-2 helicase compared to other drug molecules. The metabolism of atazanavir might decrease when used together with vapreotide; hence, detailed studies are needed for use of the combination [32]. The lack of wet laboratory experimental works on the effect of drugs on viruses is considered to be a major limitation of the study. It indicates that vapreotide is a potent inhibitor of SARS-CoV-2 helicase and may be an option for treating COVID-19 after detailed wet lab studies.
In conclusion, this study identified vapreotide as a potential drug with the lowest binding free energy on interactions with SARS-CoV-2 helicase. The designed drug selection pipeline will serve in the future for novel usage of clinically approved drugs. The prediction in the study may be beneficial to determine the best drugs to manage the fast spread of COVID-19.