Introduction
Diabetic neuropathy, one of the most common and earliest microvascular complications in type 2 diabetes, is presented as sensory, motor, and autonomic neuropathy. Diabetic sensorimotor polyneuropathy is a symmetrical, distal polyneuropathy attributable to metabolic and microvascular alterations as a result of chronic hyperglycaemia and associated metabolic changes including mitochondrial dysfunction and enhanced generation of reactive oxygen species [1, 2]. Cardiac autonomic neuropathy (CAN), perhaps the least understood complication of diabetes, is defined as the impairment of cardiovascular autonomic regulation caused by damage to the autonomic nerve fibres that innervate the heart and blood vessels [3, 4].
The development of diabetic neuropathy is a multifactorial process, although the exact pathogenic mechanism is not fully understood. The key metabolic components are the hyperglycaemia-induced activation of alternative metabolic pathways including increased polyol and hexosamine pathway flux, mitochondrial dysfunction, protein kinase C activation, the enhanced formation of reactive oxygen species, and the altered generation of endothelial nitric oxide (NO) leading to endothelial dysfunction [5–8]. The earliest and most representative findings in diabetic sensorimotor peripheral neuropathy are small-nerve fibre degeneration and endoneurial microangiopathy. During the progression of diabetic neuropathy, hyperglycaemia-induced oxidative stress, activation of alternative metabolic pathways, and decreased antioxidant protection are thought to contribute to endothelial dysfunction and decreased endoneurial blood flow with consecutive hypoxia, causing further nerve damage [5, 9]. Experimental analysis revealed that these changes in nerve microcirculation may be mediated by alterations in NO metabolism. Overproduction of superoxide anion in diabetic neuropathy leads to binding of this free radical of oxygen to NO forming highly reactive peroxynitrite, which inactivates endothelial NO synthase and triggers endothelial cell apoptosis [10, 11]. Inhibition of endothelial NO synthase by N-nitro-L-arginine resulted in decreased nerve blood flow, and this effect may be reversed by L-arginine [12].
Based on this approach, inhibition of NO synthesis may promote the progression of diabetic neuropathy via endothelial dysfunction caused by reduced endoneurial blood flow and impaired endothelium-dependent vascular relaxation. Experimental studies suggest that reduction in endoneurial blood flow precedes slowing of motor nerve conduction velocity and that the generation of reactive oxygen species could be partially responsible for the development of diabetic neuropathy [9, 13].
Asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NO synthase, is reported to increase in patients with diabetes and was suggested to play a role in the pathogenesis of accelerated atherosclerosis [14]. Previous studies documented that ADMA seems to be a predictor of the cardiovascular outcome in diabetic patients [14, 15]. Other investigators have shown that the changes in the ADMA level may contribute to modified vascular and endothelial functions in diabetic neuropathy [16].
A-lipoic acid (ALA), as a cofactor for pyruvate dehydrogenase and α-ketoglutarate dehydrogenase activity, is involved in mitochondrial oxidative metabolism and has beneficial effects on diabetic neuropathy. α-lipoic acid has been reported to improve nerve conduction velocity and protects peripheral nerves from hyperglycaemia-induced oxidative stress in experimental diabetic neuropathy [17]. ALA may improve cellular redox potential by increasing de novo synthesis of cellular glutathione, enhancing the NADPH oxidase activity of neuronal NO synthase, inhibiting nuclear factor κB activity, and improving NO-mediated vasodilation [9, 18]. Previous research has demonstrated that ADMA levels decreased after a short-term treatment with ALA in patients with type 2 diabetes [19]. However, the effects of ALA on these oxidative and endothelial markers and their correlations with the severity of cardiac autonomic and peripheral sensorimotor polyneuropathy are not fully clarified.
Therefore, the aim of our work was to assess the relationship between markers of endothelial dysfunction and NO synthesis in type 2 diabetic patients with peripheral neuropathy after six-months of ALA treatment. Moreover, we looked for associations between the changes of ADMA level and the severity of CAN and peripheral sensory neuropathy after ALA treatment.
Material and methods
Study population
Fifty-four type 2 diabetic patients with neuropathy (22 men and 32 women; mean age: 64.15 ±8.66 years; mean known type 2 diabetes duration to initiation of our study: 12.4 years [interquartile range: 4.1–14.7 years]; duration of diabetic neuropathy: 3.2 ±1.4 years) were enrolled in the study. All patients were treated daily by oral route with 600 mg ALA (WÖRWAG Pharma GmbH, Böblingen, Germany) for 6 months. Furthermore, 28 age- and gender-matched diabetic control subjects without neuropathy were also enrolled (duration of diabetes was 12.1 years [interquartile range: 4.0–14.6 years]). All patients were controlled with oral antidiabetic agents (metformin, sulfonylurea, and/or DDP4-inhibitors), and subjects on insulin therapy were excluded. Patients with a history of diabetic proliferative retinopathy, diabetic neuropathy (eGFR < 60 ml/min/1.73 m2, persistent albuminuria), or type 1 diabetes were also excluded. Also, we excluded subjects with alcoholism, known liver diseases, endocrine and autoimmune disease, haematological and neurological disorders that can be associated with peripheral neuropathy. Patients with prior cardiovascular disease, established coronary artery disease or myocardial infarction, severe congestive heart failure (NYHA class II–IV), smokers, pregnant women, and subjects with established malignancy were not enrolled in our study. All the patients were recruited from the Diabetic Neuropathy Centre of Debrecen, Department of Internal Medicine, University of Debrecen, Faculty of Medicine, Debrecen, Hungary. All participants provided written informed consent. The study protocol was approved by the local and regional ethical committees, and the study was carried out in accordance with the Declaration of Helsinki.
Blood sampling
Venous blood samples were taken after an overnight fasting, and sera were prepared immediately. Routine laboratory analyses (total cholesterol, triglyceride, high-density lipoprotein-cholesterol – HDL-C, low-density lipoprotein-cholesterol – LDL-C, glucose, haemoglobin A1c – HbA1c, creatinine, and uric acid) were performed from fresh sera with a Cobas c501 autoanalyser (Roche Ltd., Mannheim, Germany) in the Department of Laboratory Medicine, University of Debrecen, Faculty of Medicine, Debrecen, Hungary. Reagents were purchased from the same vendor, and tests were performed according to the recommendation of the manufacturer.
The sera for enzyme activity measurements and for Enzyme-Linked Immunosorbent Assay (ELISA) determinations were kept at –70°C before analysis.
ADMA measurement
Serum ADMA concentrations were measured by commercially available competitive enzyme-linked immunosorbent assay kit (ADMA-ELISA; DLD Diagnostika GmbH, Hamburg, Germany) with intra-assay coefficients of variation (CVs) ranging from 5.7% to 6.4% and inter-assay CVs ranging from 8.3% to 10.3%. Measurements of ADMA level in sera were performed according to the manufacturer’s instructions. The values were expressed as µmol/l.
TNF-α measurement
Serum levels of tumor necrosis factor-α (TNF-α) were assessed using the TNF-α (ELISA) test (R&D Systems Europe Ltd., Abington, England). Measurements of the TNF-α levels in the sera were performed according to the recommendations of the manufacturer. The intra-assay CVs ranged from 1.9% to 2.2%, and inter-assay CVs ranged from 6.2% to 6.7%. The values were expressed as pg/ml.
Measurement of oxidised low-density lipoprotein
Serum concentrations of oxidised low-density lipoprotein (oxLDL) were detected by a commercially sandwich ELISA kit (Mercodia AB, Sweden), which is based on the direct sandwich technique in which 2 monoclonal antibodies are directed against separate antigenic determinants on the oxidised apolipoprotein B molecule. The coefficient variations of the intra- and inter-assay for measurement of oxLDL were 5.5–7.3% and 4–6.2%, respectively, and the sensitivity was < 1 mU/l.
ICAM-1 and VCAM-1 measurement
The intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) levels were measured with use of human soluble ICAM-1 and VCAM-1 sandwich ELISA kits (R&D Systems Europe Ltd., Abington, England). ELISA procedures were carried out according to the manufacturer’s instructions. The intra-assay CVs and inter-assay CVs were in the range 3.7–5.2% and 4.4–6.7% (ICAM-1), and 2.3–3.6% and 5.5–7.8% (VCAM-1), respectively. The values were expressed as ng/ml.
Assay for nitrite concentration
The nitrite concentration was measured as an indicator of NO production, according to the Griess reaction [20]. Briefly, 300 µl of deprotonised plasma was incubated with an equal volume of Griess reagent (sulphanilamide and N-(1-naphtyl)ethylenediamine dihydrochloride dissolved in 2.5% H3PO4 as 0.5% and 0.05% solution, respectively, and mixed in the volume ratio 1 : 1 immediately before use) for 10 min at room temperature, in the dark. The optical density was measured spectrophotometrically at 550 nm. Nitrite concentration was determined using sodium nitrite as standard (10–100 µmol/l). The values were expressed as µmol/l.
Assessment of autonomic and peripheral nerve function
All participants underwent detailed assessment of peripheral neuropathy (DN4 questionnaire in screening for neuropathic pain syndrome, vibration perception threshold, quantitative sensory testing) and in-vivo corneal confocal microscopy by ophthalmologist for the diagnosis of diabetic sensorimotor polyneuropathy. Peripheral sensory nerve function was assessed through current perception threshold testing (CPT) using a Neurometer® (Neurotron Inc., Baltimore, Maryland, USA, 2002). Previously it was reported that this neurodiagnostic device is capable of detecting peripheral sensory neuropathy in various diseases including diabetes mellitus [21]. Neurometer CPT testing delivers a sinusoidal alternating current stimuli at 3 different frequencies: 5 Hz, 250 Hz, and 2000 Hz, assessing small unmyelinated C-fibre, small myelinated Aβ-fibre, and large myelinated Aß-fibre function, respectively. This intensity alignment is conducted to approach the sensory threshold with a ±50 microampere (µA) range from a total range of 0 to 9.99 milliamperes [22, 23]. The current stimuli were applied to dorsal surfaces of the distal phalanges of the index finger and great toe unilaterally via 2 small electrodes and the intensity was increased until the participants experienced a painless sensation. Neurometer CPT testing adjusts the level of stimulation based on the patient’s response automatically. The participants were presented with 5 to 7 randomly generated sets of stimuli above and below their level of perception and asked to choose which of the 2 stimuli felt stronger using an automated forced choice protocol. A current perception threshold value (mA) based on the minimal current perceived was calculated once a sufficient number of correct consecutive responses had been obtained.
Autonomic function was assessed by means of Ewing’s five standard cardiovascular reflex tests: changes in heart rate during deep inspiration and expiration, heart rate responses to standing up (30/15 ratio), Valsalva manoeuvre, systolic blood pressure fluctuation to standing up, and changes in diastolic pressure during a sustained handgrip [24]. A score was created to express the severity of autonomic neuropathy, based on the results of the 5 tests (normal: 0, borderline: 1, abnormal: 2). The composite autonomic score (CAS) was in the interval of 0–10. A score of 0–1 was taken as normal, 2–3 as mild, 4–6 as moderate, and 7–10 as severe CAN.
Statistical methods
Statistical analyses were performed using Statistica 13.5.0.17 software (TIBCO Software Inc., USA). Normality of distribution was tested by the Kolmogorov–Smirnov test. In the case of normal distribution, the differences between anthropometry and laboratory parameters in control and patient groups were analysed with t-test and multiple linear regression. Data were expressed as means ± SD. Spearman correlation was used to investigate the relationship between selected variables. Current perception threshold values were expressed as mean ± SD, and the Mann-Whitney U test was performed to compare the CPT values obtained from patients with those from control subjects. Spearman’s rank correlation coefficient was used to evaluate the degree of correlation between the CPT values obtained with stimuli of different frequencies. The p ≤ 0.05 probability values were considered statistically significant.
Results
Clinical and laboratory characteristics of patients before and after ALA treatment and data of control subjects are summarised in Table I.
Table I
Clinical and laboratory characteristics of patients
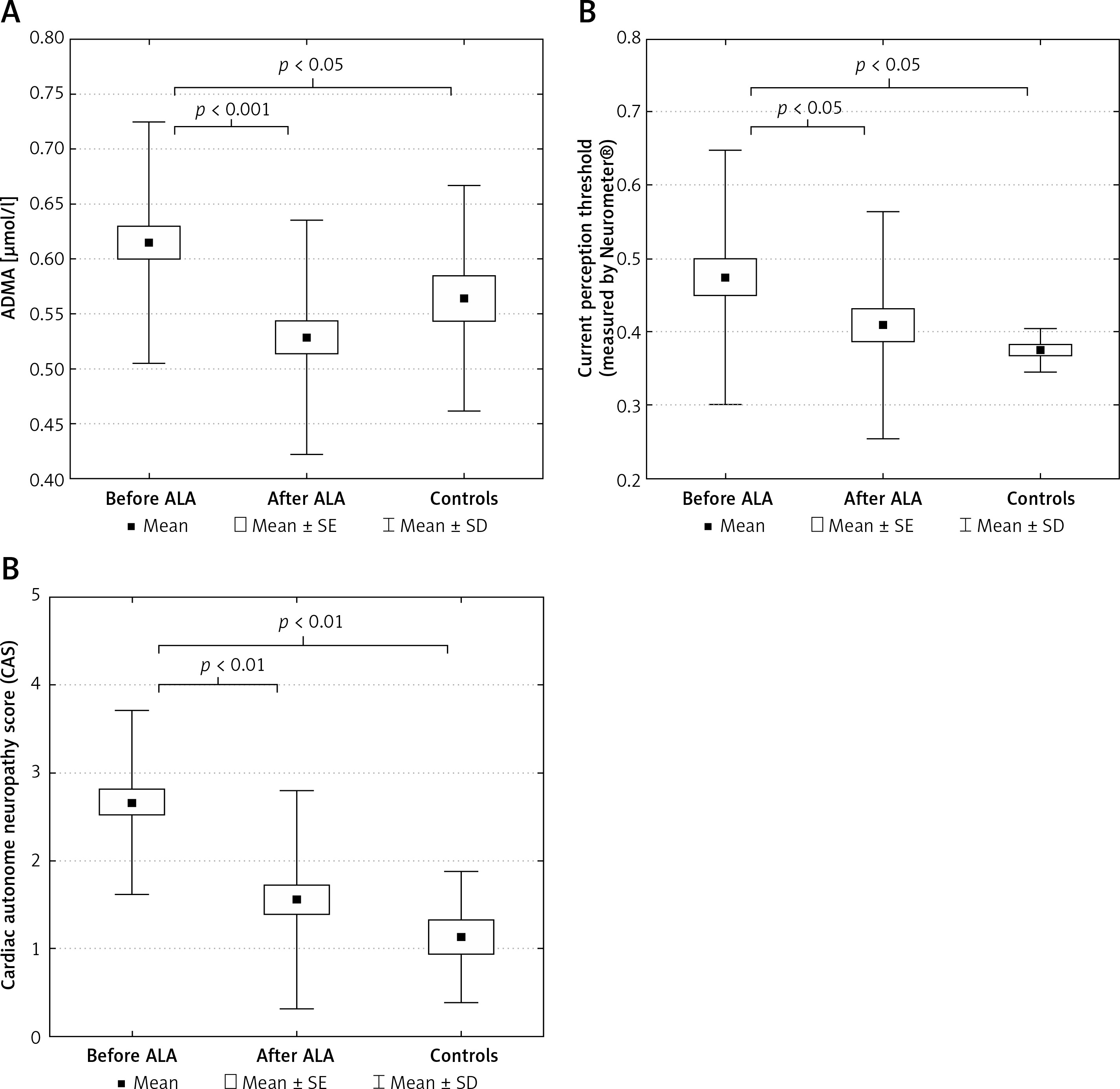
There were no significant change in body mass index, glucose, creatinine, uric acid, HbA1c, VCAM-1, ICAM-1 levels, and lipid parameters in the patient group after ALA treatment. The asymmetric dimethylarginine level significantly decreased, while the NO level increased significantly in patients after ALA treatment. The initial ADMA level was significantly higher in patients compared to controls (Table I and Figures 1 A). The level of TNF-α significantly decreased after treatment with ALA. A significant improvement of CPT measured by Neurometer CPT testing and lower CAS were detected in patients with diabetic neuropathy after receiving ALA treatment. Both CPT and CAS were higher in patients before ALA treatment compared to controls (Table I and Figures 1 B, C). The VCAM-1 level was significantly higher in patients with diabetic neuropathy both before and after ALA treatment compared to control subjects (Table I).
Figure 1
A – Asymmetric dimethyl arginine levels. B – Improvement of current perception threshold and (C) composite autonomic score in type 2 diabetic patients with neuropathy before and after α-lipoic acid (ALA) treatment and in control type 2 diabetic subjects free of ALA treatment
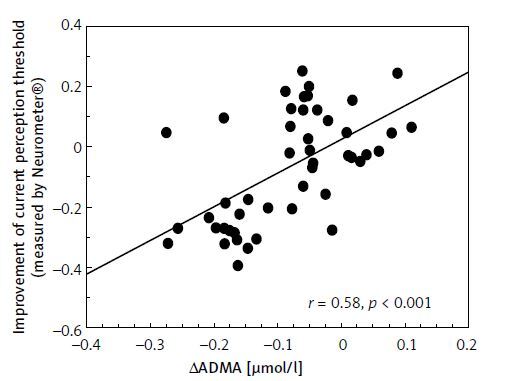
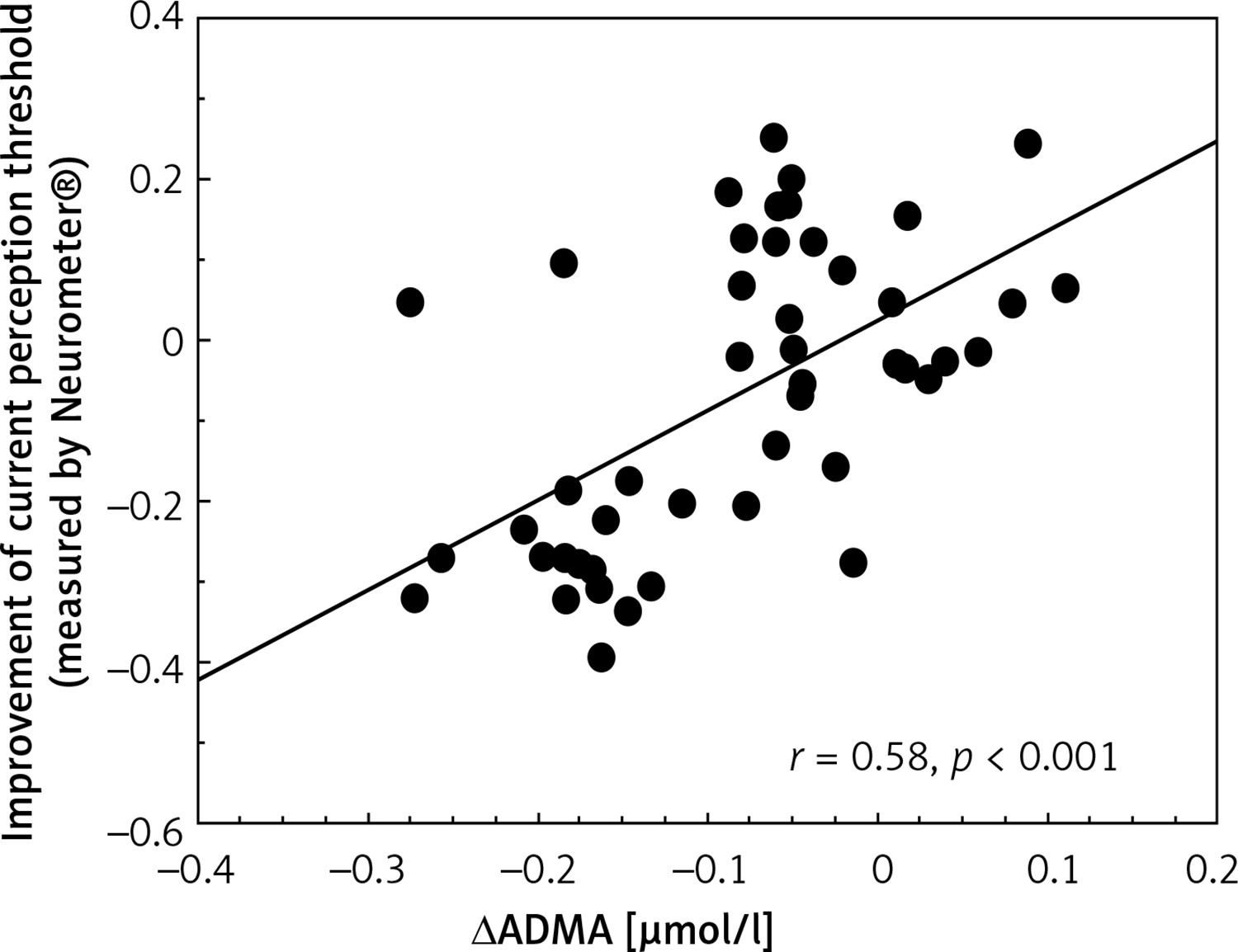
The improvement of CPT values was correlated positively with the change of ADMA levels (Figure 2). The change of TNF-α levels showed a positive correlation with the change of ADMA levels (r = 0.31, p < 0.05, not shown). The changes of ICAM-1 concentrations were correlated positively with VCAM-1 and TNF-α levels (r = 0.43, p < 0.01; r = 0.49, p < 0.01, respectively, not shown). The improvement of CPT values was correlated significantly with the decrease in CAS (r = 0.77, p < 0.001, not shown).
Figure 2
Correlation between improvement of current perception threshold measured by Neurometer and changes in asymmetric dimethyl arginine levels in type 2 diabetic patients with neuropathy
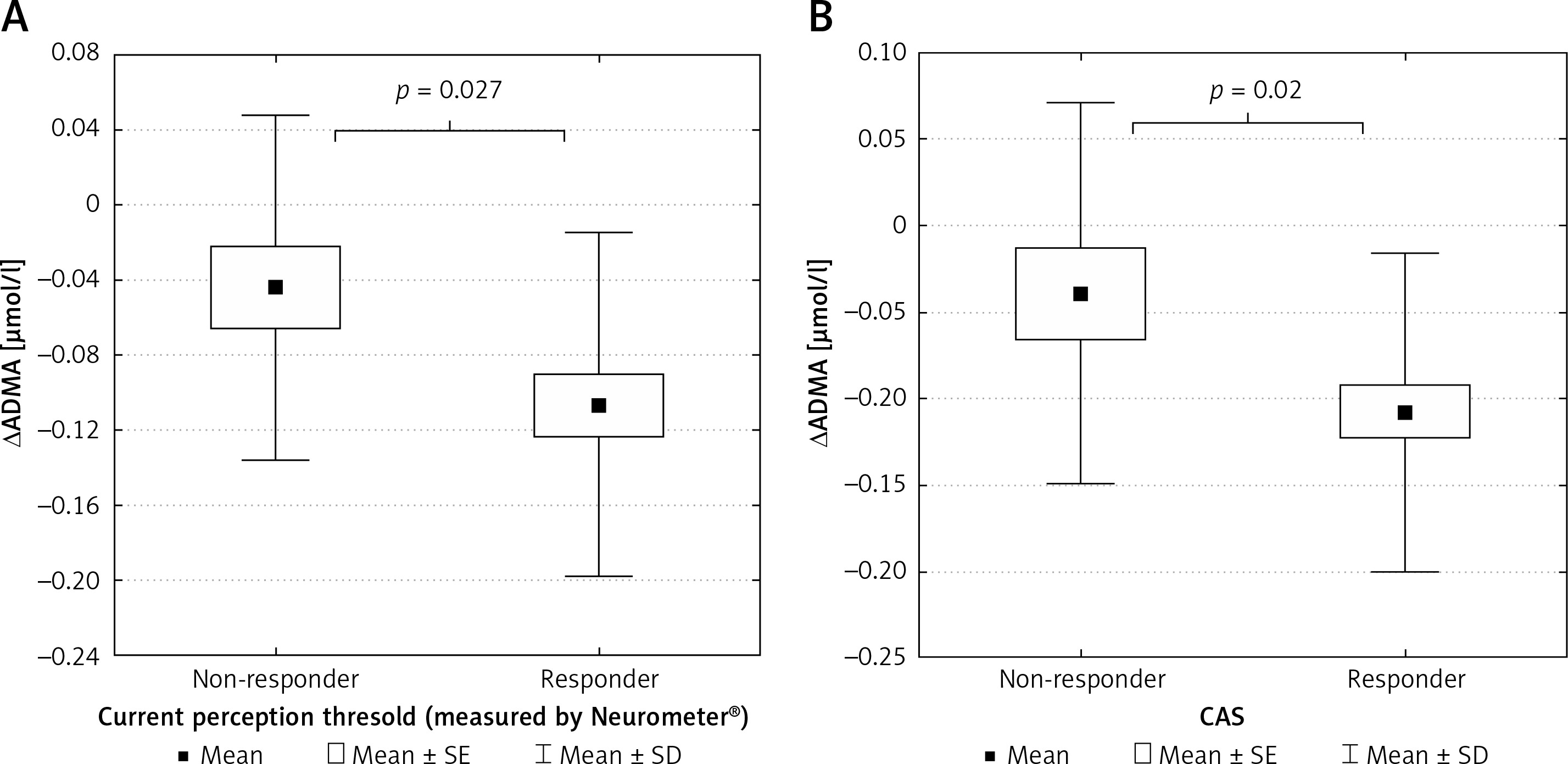
The patient group was divided into 2 subgroups according to the response to ALA treatment, defined by the clinical symptoms and the improvement in CPT and CAS. We identified 36 responders (9 male/27 female) and 18 non-responders (6 male/12 female) (Figures 3 A, B). Responders were defined as having significant changes in neuropathic pain symptoms measured by standardised Total Symptom Score and significant improvements in CPT and CAS. Decreases in ADMA levels were significantly greater in responder patients compared to non-responders identified by both CPT (p < 0.05) (Figure 4 A) and CAS (p < 0.05) (Figure 4 B).
Figure 3
Changes in (A) peripheral nerve function defined by current perception threshold measured by Neurometer and (B) composite autonomic score in non-responder and responder type 2 diabetic patients with neuropathy
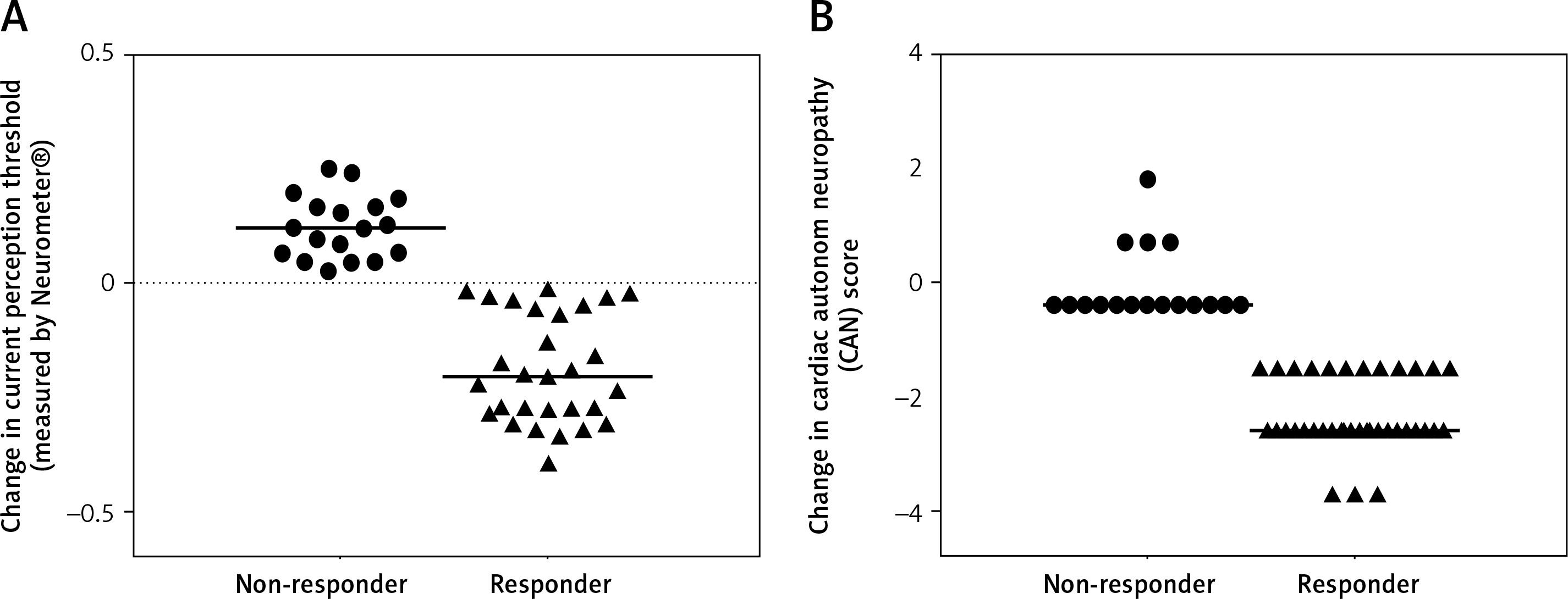
Figure 4
A – Changes in asymmetric dimethyl arginine levels in non-responder and responder type 2 diabetic patients with neuropathy defined by current perception threshold measured by Neurometer. B – Changes in asymmetric dimethyl arginine levels in non-responder and responder type 2 diabetic patients with neuropathy defined by composite autonomic score
Discussion
Despite efforts to make an early diagnosis optimal glycaemic control remains the best measure available to prevent or stop the progression of diabetic neuropathy. Chronic inflammation, increased oxidative stress, and impaired antioxidant response associated to hyperglycaemia lead to endothelial cell dysfunction and increased expression of adhesion molecules such as VCAM-1 and ICAM-1. The expression of endothelial adhesion molecules are involved in the adhesion of leukocytes and, subsequently, the progression of atherosclerosis and the occurrence of coronary heart disease [25]. Chronic inflammatory state in obesity and insulin resistance, decreased NO bioavailability, and increased VCAM-1 and oxLDL levels may also be participating factors in the vascular complications of type 2 diabetes [26, 27]. A previous in vitro study on human endothelial cells investigated the suggested role of antioxidant ALA in TNF-α induced adhesion molecule expression and NF-κB signalling. They found that ALA dose-dependently inhibited TNF-α-induced IκB kinase activation, subsequent degradation of IκB, the cytoplasmic NF-κB inhibitor, and nuclear translocation of NF-κB [28]. We also measured significantly higher VCAM-1 levels compared to controls; however, there was no significant changes in ICAM-1 and VCAM-1 levels after ALA treatment among diabetic patients. To date, there are no data on the effect of ALA on serum levels of VCAM-1 and ICAM-1 in diabetic patient populations. In a previous in vitro study, ALA suppressed the homocysteine-stimulated ICAM-1 and VCAM-1 expression of human aortic endothelial cells [29]. Elevated levels of TNF-α are also associated with coronary artery disease via inhibition of NO-mediated dilation of coronary arterioles and subsequent production of superoxide in endothelial cells [30]. Although serum NO levels were higher in our patients compared to controls, the difference was not significant.
Asymmetric dimethylarginine, an endogenous inhibitor of endothelial NO synthase, induces expression of adhesion molecules in endothelial cells, oxLDL in vascular endothelium, and promotes the adhesion of thrombocytes to the endothelium [31]. Various studies documented that ADMA seems to be not only a cause of endothelial dysfunction [32], but also a predictor of the cardiovascular risk in type 2 diabetes [14, 15]. ADMA has been closely related to insulin resistance and elevated ADMA levels demonstrated in type 2 diabetic patients with neuropathy, suggesting a role in the development of diabetic neuropathy [33, 34]. Other investigators have found higher ADMA levels in diabetic patients compared to non-diabetic controls; however, the changes in ADMA concentrations have not shown a consistent relationship in subjects with or without diabetic neuropathy [16]. In line with these previous data, we also found significantly higher ADMA levels in our patients with neuropathy before ALA treatment. Indeed, because ADMA is mainly metabolised by dimethylarginine dimethylaminohydrolase (DDAH), it is conceivable that the inhibition of ADMA via up-regulation of DDAH may be a novel therapeutic target for the prevention of chemical vapour deposition in patients with diabetes [35].
Multiple clinical trials have proven the pathogenetically oriented, disease-modifying effect of ALA treatment. According to these studies, significant improvements in neurological function, affecting both sensorimotor and autonomic components of the peripheral nervous system [36, 37], and in components of the CAN [38], were found in patients with diabetic neuropathy. Our results support the beneficial effect of ALA therapy on symptoms of peripheral neuropathy. The mechanism of improvement of neuropathic symptoms and nerve function is thought to be related to improvement in endothelial dysfunction.
In our study we have found significantly decreased ADMA levels and elevated NO concentration as a result of ALA treatment in type 2 diabetic patients. Moreover, we have found significantly lower serum TNF-α levels after 6-month treatment. These data suggest that ALA might modulate the activity of NO synthase involved in increasing endoneurial blood flow and decreasing the expression of endothelial adhesion molecules. This beneficial effect may be due to improved antioxidant status and endothelial function in diabetic neuropathy.
Although several authors have demonstrated decreased ADMA concentration in type 2 diabetic patients with diabetic nephropathy and neuropathy receiving ALA treatment for 6–12 weeks [19, 39], there was no previous study focusing on the changes of ADMA levels after long-term therapy. According to these studies, ADMA was found to be an independent risk factor for cardiovascular outcome in diabetic nephropathy, and ALA treatment may be associated with cardiovascular risk reduction, in part by decreasing the plasma level of ADMA [19, 39]. Asymmetric dimethylarginine inhibits the endothelial NO synthase, which may explain partly the impaired endothelial function and reduced endoneurial blood flow [40]. Our findings indicate a possible close association between chronic inflammation, increased endothelial ADMA concentrations, and a decreased antioxidant activity in response to increased production of ROS in the pathogenesis of neuropathy in type 2 diabetes. We have found a significant positive correlation between the changes of TNF-α and ADMA levels after 6-month ALA treatment. The same correlation was detected between the changes of serum TNF-α and ICAM-1 concentrations in our study. Based on these data, we assume that the elevated ADMA levels and oxidative stress may be responsible for endothelial dysfunction in diabetic neuropathy. However, supplementation with ALA may have a protective effect against diabetic nerve damage by restoring endothelial function and decreasing the production of the inflammatory cytokine TNF-α in type 2 diabetes. Our results support the initial hypothesis that increased ADMA level may be a marker of endothelial dysfunction and cardiovascular outcome in diabetic neuropathy. The level of ADMA correlated with the severity of peripheral sensory neuropathy and significantly lower ADMA concentration was detected in patients with decreased CAS after ALA treatment. Therefore, changes in serum ADMA levels may predict the clinical response to ALA treatment.
There are some limitations of the study. The power of the study may be reduced because of the relatively small number of patients. Data on other markers of endothelial dysfunction, such as P- and E-selectin, von Willebrand factor, plasminogen activator inhibitor-1, and monocyte chemoattractant protein-1, as well as flow mediated dilatation and arterial stiffness parameters, would improve our knowledge about the effect of ALA treatment on endothelial dysfunction and its contribution to the beneficial effects on cardiac and peripheral neuropathy. Evaluation of sural nerve automated nerve conduction in the diagnosis of peripheral neuropathy could give useful additional information [41].
In conclusion, our results highlight the significant role of ALA administration in the long-term treatment of peripheral and CAN. Monitoring of ADMA serum level may predict the efficacy of ALA treatment detected by Neurometer CPT testing. Further studies are needed to clarify the additional favourable effects of ALA in neuropathy. Data on other endothelial biomarkers may improve our knowledge about the effect of ALA treatment on endothelial dysfunction. However, the positive effect of ALA on endothelial function through the severity of peripheral and CAN may underline the importance of oxidative stress in the pathomechanism of diabetic neuropathy and enhances the development of novel antioxidant therapeutic agents.