Introduction
Ischemia-reperfusion (IR) injury is an unavoidable event in some clinical settings such as kidney surgery and is one of the main causes of acute kidney injury (AKI) after transplantation. IR injury is associated with multiple processes, including free reactive oxygen species (ROS) formation, inflammatory response activation, and endothelial dysfunction, which all contribute to post-ischemic renal failure [1, 2]. Therapeutic methods to reduce renal IR injury have been reviewed recently by McCafferty et al. [3]. Pharmacological intervention [4–6] and ischemic conditioning [3] are the two most applicable and effective therapeutic methods to reduce ischemic injury in different organs.
Remote ischemic conditioning (RIC), which is triggered by applying short-time cyclic ischemia and reperfusion in distant organs or limbs, seems to be the most practically efficient strategy against renal IR injury. The RIC can be applied before the onset of prolonged ischemia, which is termed remote preconditioning (RPC), or at the onset of reperfusion, which is defined as remote post-conditioning (RPOC). These two methods have some limitations. In the case of RPIC, its application requires foreknowledge of the ischemic event, and RPOC is mostly applicable to patients undergoing manual reperfusion.
A novel alternative approach to RIC, remote per-conditioning (RPEC), was first reported by Schmidt et al. in 2007 and is achieved by short-time IR cycles in remote organs or limbs during target organ ischemia [7]. RPEC is different from RPC and RPOC in terms of time onset in relation to the main ischemic insult. This method has the advantage of being practical even after the onset of the ischemic insult. The potent renoprotection and hepatoprotection of this procedure were reported by us in an animal model of renal IR injury [8, 9].
Identification of the mechanisms responsible for remote per-conditioning is important, not only for the understanding of the pathophysiology of its protection but also for formulating therapeutic strategies aimed at mimicking the protective mechanisms in pharmacological manipulation. However, RPEC is a newly described intervention in which it is not clear how these protective mechanisms are afforded by the short-time cycles of IR.
Several mediators have been proposed to be involved in the protective effects of ischemic conditioning methods, such as adenosine, nitric oxide (NO), and reactive oxygen species. Nitric oxide is known to participate not only in the normal functions of the kidney but also in the pathophysiology of renal ischemic injury [10]. Nitric oxide is synthesized by one of the nitric oxide synthase (NOS) enzyme isoforms. The expression of neuronal NOS is mostly limited to neural tissues. Inducible nitric oxide synthase (iNOS) is not expressed under normal circumstances but is up-regulated during inflammatory conditions in renal epithelial cells, neutrophils, and T lymphocytes. Endothelial nitric oxide synthase (eNOS) is constitutively expressed in many cell types, including kidney endothelial cells and epithelial cells. There are studies indicating that NO donors protect the kidney from ischemic injury [11]. Moreover, inhibition of NO synthesis increases susceptibility of the kidney cells to IR injury [12]. By contrast, there are other studies demonstrating that inhibition of NOS protects organs against ischemic damage [13]. However, the role of iNOS and eNOS enzymes in the protective effect of the newly described method, RPEC, has not been studied.
Recently, we found that the protection afforded by RPEC may be a consequence of reductions in lipid peroxidation, attenuation of pro-inflammatory molecules, and intensification of anti-oxidant systems [14, 15]. In the present study, first, we looked for the possible involvement of NO by investigating the effects of induction of RPEC on the expression of iNOS and eNOS after renal IR. Secondly, we considered the importance of eNOS and iNOS in the modulation of RPEC by investigating renal functional and histological changes, and oxidative and pro-inflammatory mechanisms in animals treated with NOS inhibitors.
MATERIAL AND METHODS
Animal preparation
Adult male Sprague-Dawley rats (220–270 g) were maintained on a standard chow pellets and water ad libitum in a room under normal lighting conditions. All animal experimental protocols were approved by the Animal Ethics Committee of Tehran University of Medical Sciences.
Animals’ anesthesia was achieved by sodium pentobarbital (60 mg/kg, i.p.). Heart rate and systolic blood pressure were monitored by a tail-cuff connected to a pulse transducer device (MLT125/R; ADInstruments, Castle Hill, NSW, Australia). The transducer was connected to a Power-Lab/4SP data-acquisition system (Chart, version 5; ADInstruments). Rats with hypotension during the surgery were excluded from the study.
Technique of hind limb per-conditioning
A longitudinal skin incision was made over the right antero-medial thigh. The femoral artery was isolated from the vein, nerve and surrounding muscles and was clamped. Five minutes of limb ischemia followed by 5 min of reperfusion (4 cycles) was used as the RPEC technique.
Experimental protocol
Rats were randomly assigned to the following five groups (n = 7): (1) sham-operated group (sham): underwent sham surgery without ischemia; (2) Ischemia/reperfusion group (IR): the left renal pedicle was occluded with a bulldog clamp for 45 min followed by 24 h reperfusion; (3) Remote per-conditioning group (RPEC): four 5-min IR cycles of left femoral artery were administered at the beginning of renal ischemia; (4) RPEC + L-NAME (L-NAME group): L-NAME (10 mg/kg, a non-specific NOS inhibitor) given intraperitoneally 30 min before renal ischemia to RPEC-treated animals; (5) RPEC + 1400W (1400w group): 1400W (1 mg/kg, a specific iNOS inhibitor) given intraperitoneally 30 min before renal ischemia to RPEC-treated animals. A sham-operated L-NAME group was used (n = 3) in which 10 mg/kg L-NAME was injected intraperitoneally 30 min before nephrectomy.
Animals of all groups underwent right nephrectomy through a flank incision at the beginning of surgery. After 45 min of left renal ischemia, the clamp was removed to allow reperfusion, which was confirmed visually. The incision was closed, animals were housed in metabolic cages and urine samples (24 h) were taken. Blood samples were drawn from the inferior vena cava and plasma was separated by centrifugation. These samples were used for measurements of renal functional parameters. Kidneys were harvested and then washed and dissected in normal saline. Part of the kidney was fixed in 10% formalin for histological evaluation, and the rest was stored at –70°C until further use.
Renal functional assessment
Blood urea nitrogen (BUN), serum and urinary creatinine, and serum and urinary Na+ were measured by colorimetric methods (Hitachi 704 autoanalyser; Hitachi, Tokyo, Japan). Moreover, urine samples were collected at the end of the 24 h reperfusion period and total volume determined. Urinary creatinine was used in conjunction with serum creatinine concentration and urine flow to calculate creatinine clearance (CCr) using standard formulas. Creatinine clearance was used as an indicator of glomerular function. Urinary Na+ was used in conjunction with serum Na+ to estimate the fractional excretion of Na+ (FENa+) using standard formulas. The FENa+ was used as an indicator of tubular function.
Activity of urinary N-acetyl-β-D-glucosaminidase (NAG), a specific indicator of tubular damage, was assessed based on the enzymatic hydrolysis of p-nitrophenyl-N-acetyl-glucosaminide at pH 4.4 and the subsequent evaluation of delivered p-nitrophenol at 405 nm by spectrophotometry [16].
Neutrophil gelatinase-associated lipocalin (NGAL) as one of the most dramatically up-regulated genes in the renal tissues after ischemia and a sensitive biomarker for AKI was assessed by quantitative real-time RT-PCR (described later).
Renal oxidative stress assessment
Malondialdehyde (MDA) in tissue samples was evaluated by the measurement of thiobarbituric acid-reactive substance (TBARS) levels at 532 nm. It was done according to the methods of Esterbauer and Cheeseman [17]. Results were expressed as μmol/100 mg tissue.
Renal tissue superoxide dismutase (SOD) activity was determined according to the method of Paoletti and Mocali [18]. In this assessment, the superoxide anion is generated from O2 in the presence of EDTA, mercaptoethanol and manganese (II) chloride. Oxidation of NADPH is related to the levels of superoxide anions in the medium.
The activity of catalase (CAT) in the renal tissues was determined according to the method of Aebi [19]. The decomposition of H2O2 was assessed using a spectrophotometer at 240 nm. Activity is indicated as a K rate constant of the first-order reaction per gram of tissue.
Renal tissue total glutathione (GSH) levels were assayed according to the method of Tietze [20] as modified by Griffith [21]. The continuous reduction of 5,5-dithiobis 2-nitrobenzoic acid as a chromogen was determined at 412 nm. The amount for each sample was extracted from the standard curve and is expressed in μmol/g of tissue.
Western blot analysis of COX-2 and TNF-α
The expression of TNF-α and COX-2 in renal tissues was evaluated using western blot analysis. Protein extracts were obtained by homogenizing samples in ice-cold lysis buffer, followed by two cycles of centrifugation at 15700xg for 15 min at 4°C. Protein concentrations were determined by the Bradford assessment. An equal amount of protein from each sample was electrophoresed on 12% SDS polyacrylamide gels and then blotted onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked in 2% skim milk prepared in TBST (Tris-buffered saline and 0.1% Tween 20) for 75 min at room temperature and then incubated overnight at 4°C with COX-2 (ABR-Affinity BioReagents, Golden, CO, USA) and TNF-α (Cell Signaling, Danvers, MA, USA) antibodies. Membranes were then washed three times with TBST, incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling, Danvers, MA, USA) for 60 min at room temperature and finally visualized using an enhanced chemiluminescence detection system (GE Healthcare, Amersham, UK). Equal loading was confirmed by stripping the immunoblots and reprobing with β-actin rabbit monoclonal antibody (Cell Signaling). At the end, the COX-2 and TNF-α protein bands were assessed semiquantitatively by densitometric evaluation of scanned X-ray films using ImageJ software, and normalized against β-actin bands.
Quantitative real-time RT-PCR analysis of mRNA expression of eNOS, iNOS and IL-6
Part of the renal tissue was placed in RNA later solution (Sigma) immediately after retrieval and stored at –70°C. For analysis of gene expression, total RNA was isolated from renal tissues using the RNeasy Mini kit (Qiagen, Valencia, CA), according to the manufacturer’s instructions. RNA quality was assessed using NanoDrop (Thermo Scientific). Complementary DNA (cDNA) was prepared by reverse transcription of total RNA using RevertAid M-MuLV enzyme in the reaction mixture according to the manufacturer’s protocol (Vivantis, Surrey, UK). Quantitative, real-time RT-PCR (qRT-PCR) with SYBR Green fluorescence detection was performed using an ABI Prism 7000 ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Carlsbad, CA) with specific primers. Sequences of primer pairs were as follows:
iNOS: F: 5′-TGCTAATGCGGAAGGTCATC; R: 5′-GC-TTCCGACTTTCCTGTCTCA,
eNOS: F: 5′-GACTTTTAAGGAAGTAGCCAATG; R: 5′-GGGGTCATAGGAGTCATTGG,
IL-6: F: 5′-AAGTCCGGAGAGGAGACTTCA; R: 5′-GCCATTGCACAACTCTTTTCTCATT,
NGAL: F: 5′-GCAGTGGCCTGATGGTTCA; R: TGATTCTCTGGCAACAGGAAA,
GAPDH: F: 5′-TGGCCTCCAAGGAGTAAGAAAC; R: 5′- GGCCTCTCTCTTGCTCTCAGTAT.
The mRNA levels of various genes were calculated after normalizing with the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All data were normalized against the GAPDH mRNA levels and expressed as fold increases relative to control ± SEM.
Histological assessment
Formaldehyde-fixed paraffin sections of kidney were stained with hematoxylin and eosin (H&E). Histopathology for all kidneys was reported per section in at least 10 randomly selected non-overlapping fields. Histological evaluation and grading of kidney damage included tubular necrosis, tubular dilatations and loss of brush borders, as well as the formation of casts and luminal debris.
Statistical analysis
Data are expressed as the mean ± SEM. Statistical analyses were performed with SPSS software version 11.5 (SPSS, USA) and GraphPad Prism version 5.01 (GraphPad, USA). One-way ANOVA was used for the comparison among the groups of data and individual group means were compared using Tukey’s multiple group comparison tests. P-values of < 0.05 were considered to be significant.
RESULTS
Effect of eNOS or iNOS inhibition on the glomerular functional protection by RPEC treatment
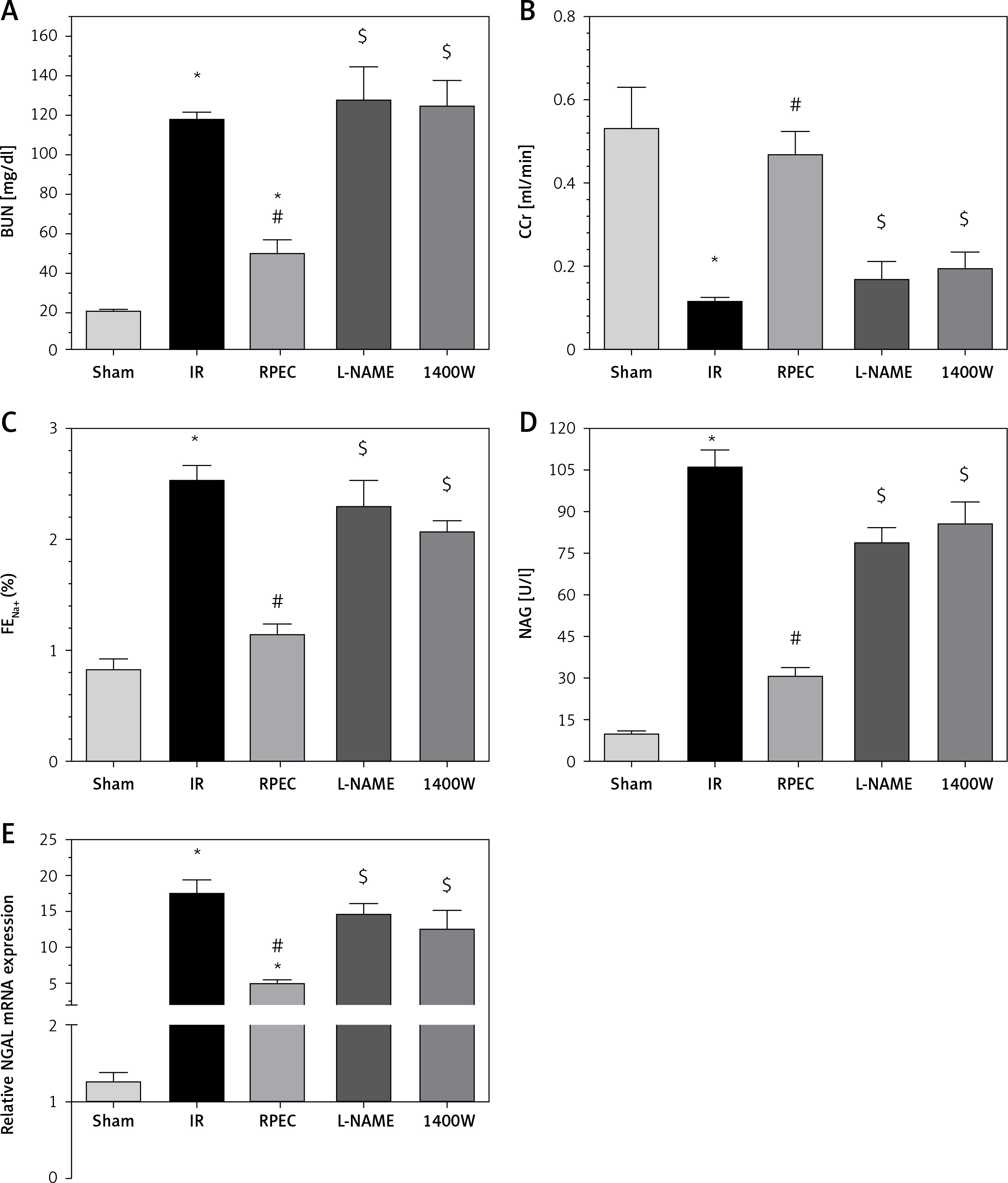
The ameliorative effect of RPEC on glomerular function of rats subjected to 45 min ischemia was completely inhibited by pretreatment with L-NAME or 1400W. As compared with RPEC rats, L-NAME and 1400W groups showed significant decreases in creatinine clearance (CCr) (Figure 1 A).
Figure 1
Effects of eNOS or iNOS inhibition on the renal functional protection by RPEC treatment. A – Blood urea nitrogen (BUN), B – measurement of creatinine clearance (CCr), C – fractional excretion of Na+ (FENa+), D – urinary N-acetyl-β-D-glucosaminidase (NAG) levels, E – neutrophil gelatinase-associated lipocalin (NGAL) expression at 24 h after reperfusion
IR – ischemia/reperfusion group, RPEC – remote pre-conditioning group, L-NAME – nonspecific NOS inhibition + RPEC, 1400W – specific iNOS inhibition + RPEC. Each column and bar represent the mean ± SEM (n = 7). *P < 0.05 compared with the sham group; #p < 0.05 compared with the IR group; $p < 0.05 compared with the RPEC group.
Effect of eNOS or iNOS inhibition on the tubular functional protection by RPEC treatment
The ameliorative effect of RPEC on tubular function of rats subjected to 45 min ischemia was completely inhibited by pretreatment with L-NAME or 1400W. As compared with RPEC rats, L-NAME and 1400W groups showed significant increases in FENa and urinary NAG levels (Figures 1 B, C).
Effect of eNOS or iNOS inhibition on the renal functional protection by RPEC treatment
The ameliorative effect of RPEC on renal function of rats subjected to 45 min ischemia was completely inhibited by pretreatment with L-NAME or 1400W. As compared with RPEC rats, L-NAME and 1400W groups showed significant increases in BUN levels as well as an increase in NGAL gene expression (Figures 1 D, E).
Effect of L-NAME on renal function in the sham-operated group
Intraperitoneally injected L-NAME sham animals showed no significant difference in BUN or creatinine (28.31 ±1.20 mg/dl and 0.80 ±0 mg/dl) from those of the normal sham group (21.00 ±1.15 mg/dl and 0.65 ±0.15 mg/dl).
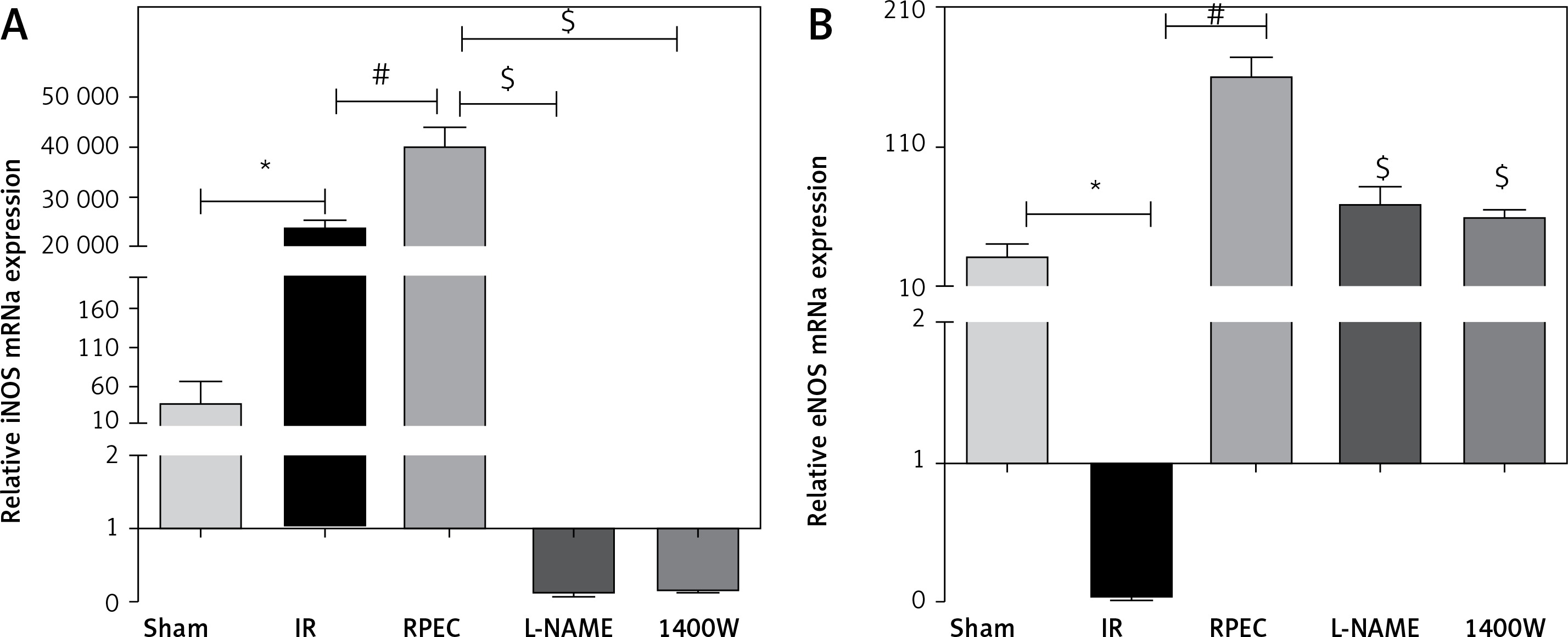
Effect of RPEC on eNOS and iNOS expression in the kidney
Renal eNOS gene expression of rats subjected to 45 min ischemia and 24 h reperfusion (IR group) showed significant decreases compared with sham-operated rats. eNOS expression was significantly higher in the RPEC group compared with the IR group. In addition, renal iNOS gene expression of the IR group showed significant increases compared with the sham group. However, iNOS expression was significantly higher in the RPEC group compared with the IR group. L-NAME and 1400W significantly reduced eNOS and iNOS expression compared with the RPEC group (Figure 2 A).
Figure 2
Effect of RPEC on iNOS (A) and eNOS (B) expression in the kidney. Measurement of renal eNOS and iNOS gene expression at 24 h after reperfusion
IR – ischemia/reperfusion group, RPEC – remote per-conditioning group, L-NAME – nonspecific NOS inhibition + RPEC, 1400W – specific iNOS inhibition + RPEC. Each column and bar represent the mean ± SEM (n = 7). *P < 0.05 compared with the sham group; #p < 0.05 compared with the IR group; $p < 0.05 compared with the RPEC group.
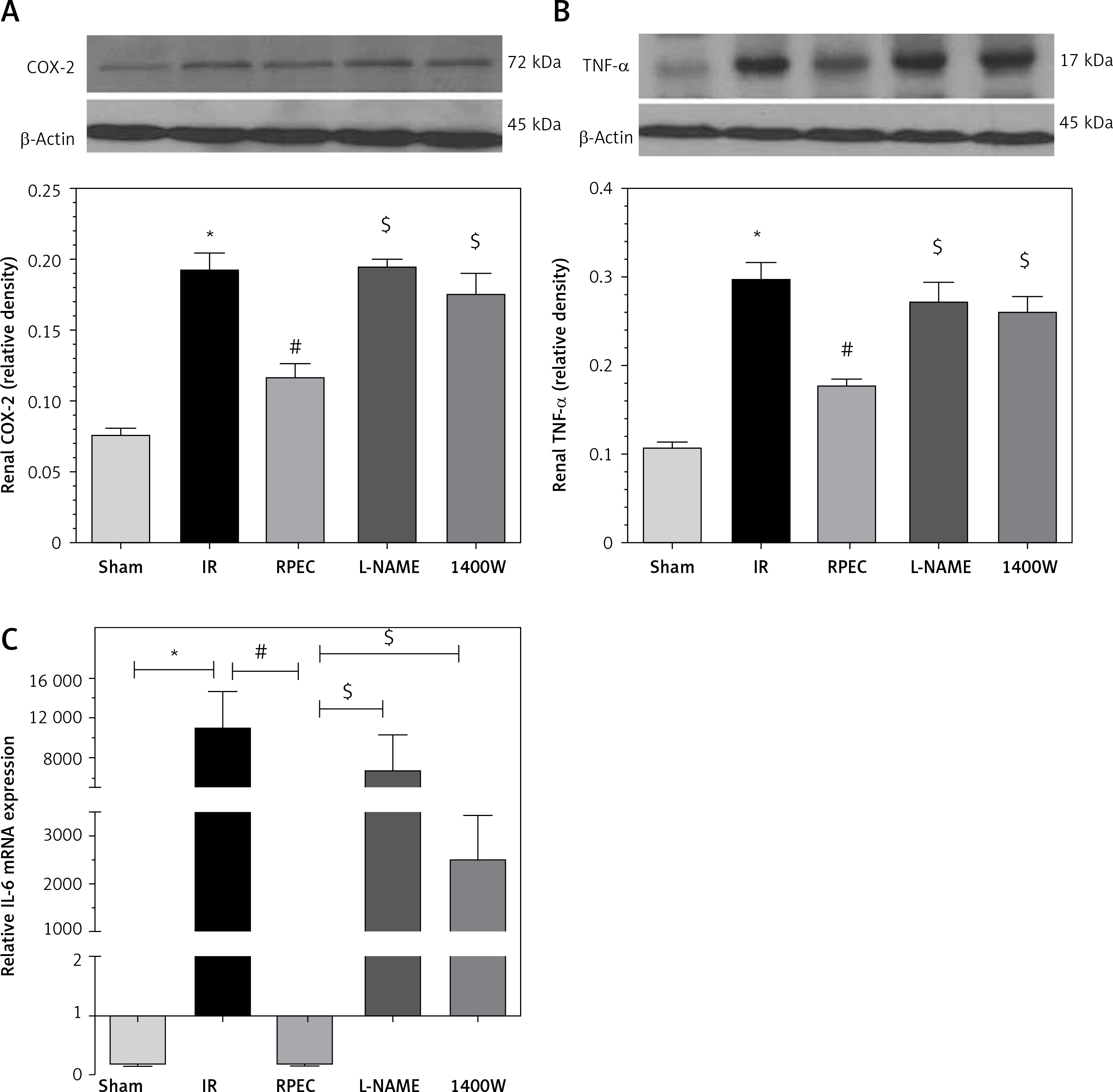
Effect of RPEC on renal TNF-α, IL-6 and COX-2 expression in the presence of NOS inhibitors
The ameliorative effect of RPEC on pro-inflam-matory marker expression of rats subjected to 45 min ischemia was completely inhibited by pretreatment with L-NAME or 1400W. As compared with RPEC rats, L-NAME and 1400W groups showed significant increases in TNF-α and COX-2 protein expression. In addition, the suppressive effect of RPEC treatment on IL-6 gene expression was abolished by L-NAME and 1400W prescription (Figure 3).
Figure 3
Effects of eNOS or iNOS inhibition on the decreased COX-2 (A), TNF-α (B) and IL-6 (C) expression by RPEC treatment. Measurement of renal TNF-α and COX-2 protein expression, and IL-6 gene expression
IR – ischemia/reperfusion group, RPEC – remote pre-conditioning group, L-NAME – nonspecific NOS inhibition + RPEC, 1400W – specific iNOS inhibition + RPEC. Each column and bar represent the mean ± SEM (n = 7). *P < 0.05 compared with the sham group; #p < 0.05 compared with the IR group; $p < 0.05 compared with the RPEC group.
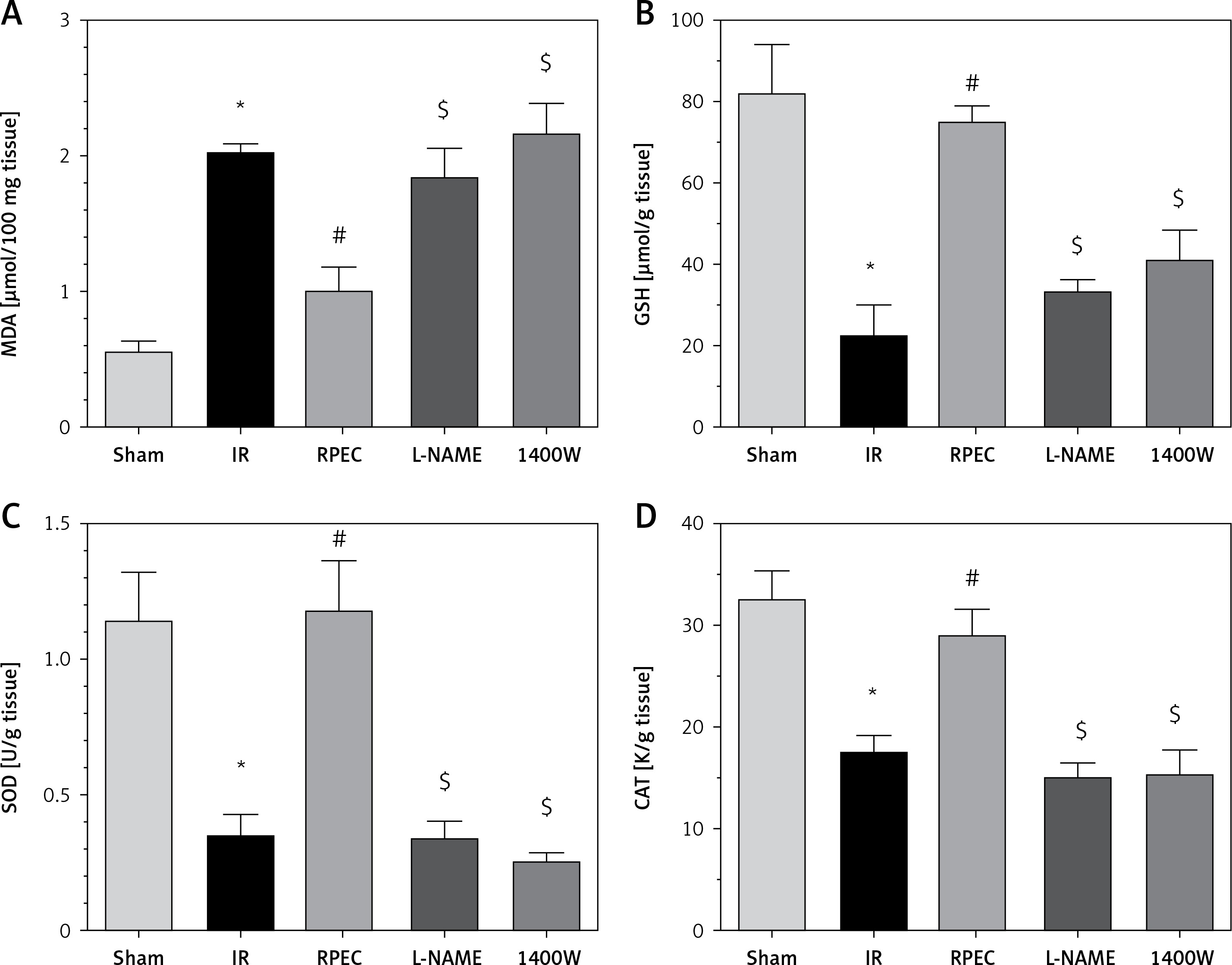
Effects of eNOS or iNOS inhibition on the renal oxidative protection by RPEC treatment
The ameliorative effect of RPEC on renal oxidative state of rats subjected to 45 min ischemia was completely inhibited by pretreatment with L-NAME or 1400W. As compared with RPEC rats, L-NAME and 1400W groups showed significant decreases in SOD and CAT activities as well as decreases in the GSH concentration and increases in MDA levels (Figure 4).
Figure 4
Effects of eNOS or iNOS inhibition on the renal oxidative protection by RPEC treatment. Measurement of renal malondialdehyde (MDA) content (A), total glutathione (GSH) levels (B) and superoxide dismutase (SOD) (C) and catalase (CAT) activity (D)
IR – ischemia/reperfusion group, RPEC – remote per-conditioning group, L-NAME – nonspecific NOS inhibition + RPEC, 1400W – specific iNOS inhibition + RPEC. Each column and bar represent the mean ± SEM (n = 7). *P < 0.05 compared with the sham group; #p < 0.05 compared with the IR group; $p < 0.05 compared with the RPEC group.
Effects of eNOS or iNOS inhibition on the renal morphological protection by RPEC treatment
The ameliorative effect of RPEC on renal histopathological changes of rats subjected to 45 min ischemia was completely inhibited by pretreatment with L-NAME or 1400W. As compared with RPEC rats, L-NAME and 1400W groups showed extensive changes in renal tissues. The changes observed in the L-NAME or 1400W groups were included severe destruction of tubules as a result of cellular necrosis, especially in the proximal tubules, cast formation and obstruction of the more distal tubules, loss of brush borders and a flattening and attenuation of tubular patency (Figure 5).
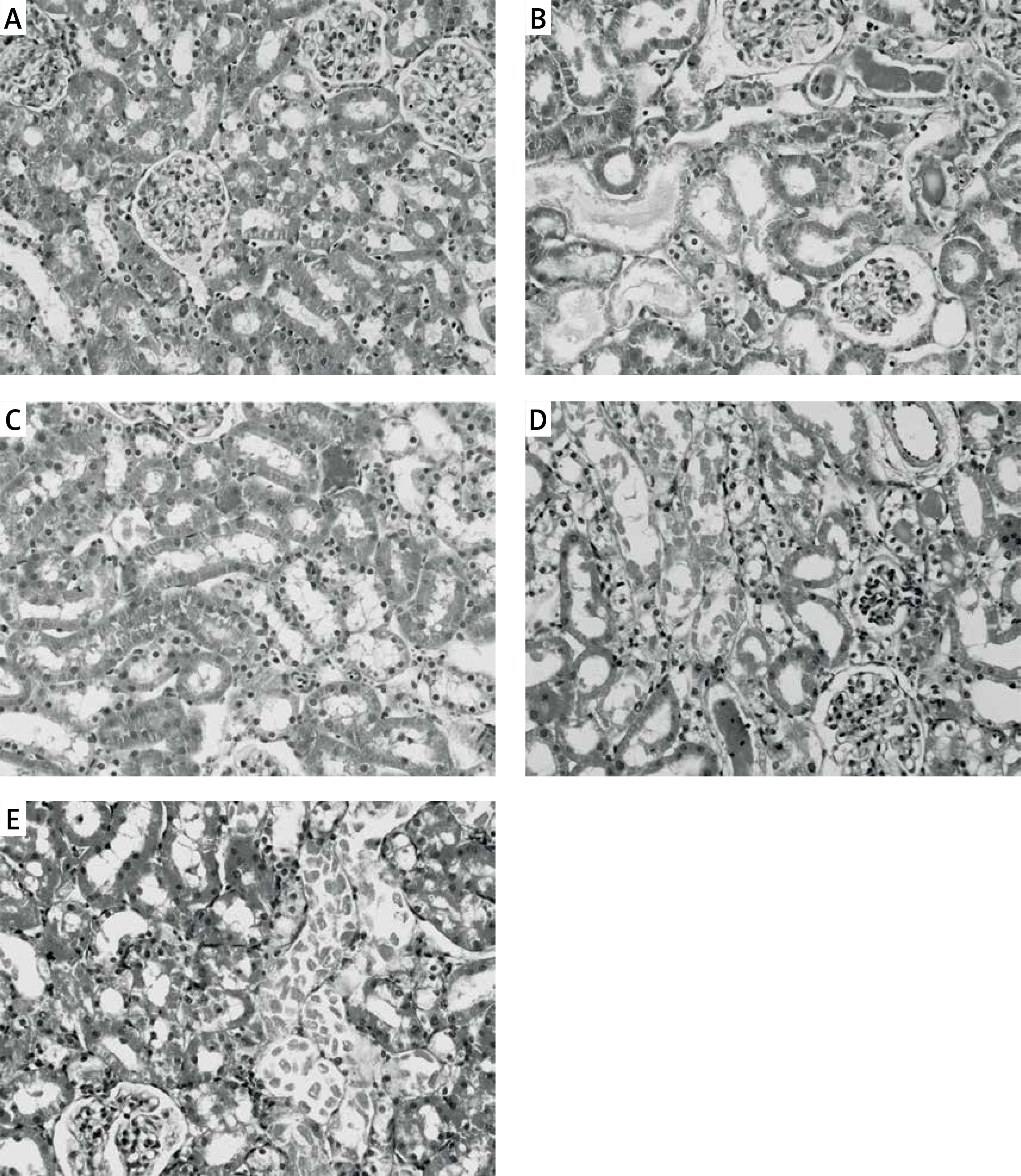
Figure 5
Effects of eNOS or iNOS inhibition on the renal morphological protection by RPEC treatment. A – sham, B – IR, ischemia/reperfusion group, C – RPEC, remote per-conditioning group, D – L-NAME, nonspecific NOS inhibition + RPEC, E – 1400W, specific iNOS inhibition + RPEC. Hematoxylin and eosin-stained sections were evaluated by light microscopy
Discussion
There is a great need for effective clinical strategies to attenuate IR injury resulting from renal surgery and transplantation. One of these strategies may be the newly described method, remote per-conditioning (RPEC). This treatment was shown to protect the kidney from ischemia reperfusion injury in our recent study [15]. However, the present study was performed to determine whether the eNOS/iNOS system is responsible for RPEC-mediated renal protection. We obtained evidence that an RPEC-mediated improvement on post-ischemic renal injury at 24 h after reperfusion may be mediated through NO pathway.
We demonstrated here that pretreatment with L-NAME, a nonselective NOS inhibitor, and 1400W, an iNOS inhibitor, significantly attenuated the renal functional protection afforded by RPEC in a rat model of renal IR. This conclusion is supported by the following specific, key findings: 1400W and L-NAME opposed the RPEC-mediated changes in (a) serum levels of urea, creatinine and creatinine clearance, (b) fractional excretion of Na+ (FENa+), urinary concentrations of NAG, and (c) renal gene expression of NGAL. L-NAME and 1400W also significantly worsened the histological evidence of IR-mediated tubular injury, which was attenuated by RPEC treatment. Moreover, L-NAME and 1400W substantially opposed the RPEC-mediated decreases in oxidative state, which was indicated by decreased: (a) SOD and CAT activities, (b) GSH levels, (c) as well as increased MDA levels in the kidney. Finally, both L-NAME and 1400W significantly inhibited the RPEC-mediated reduction in the expression of pro-inflammatory (TNF-α and IL-6) mediators and COX-2 in the kidney. The results also demonstrated that eNOS and iNOS gene expression was up-regulated in the ischemic remote per-conditioned kidneys after 24 h reperfusion. Thus, it seems that the eNOS/iNOS system plays a role in the RPEC protection against IR-induced renal injury.
There are several studies demonstrating that chronic administration of L-NAME (1 week up to a few weeks) to normal rats exerts injurious effects on the kidney associated with an increase in oxidative stress, increased local (renal) angiotensin II [22] and increased sympathetic activity [23]. In this study, we used a single dose of L-NAME (described as a reversible, non-specific inhibitor of NOS) to block NO production in the RPEC-treated animals. To exclude the pure effect of L-NAME on the non-ischemic animals, we used an L-NAME sham group. It seems that a single dose of L-NAME has no significant effect on renal functional indices after 24 h in the L-NAME sham group compared to the normal sham group. This is in line with the report of Wu et al. [24].
Nitric oxide is shown to be a mediator/modulator of IR injury in many organs, including heart and kidneys [12, 25]. However, there are still controversies concerning the actual protective effects of NO. In a review by Phillips et al. in 2009 the positive role of NO was emphasized and suggested that the cellular effects of NO depend on its concentration, site of release and duration of action. These can be as a result of experimental situations such as the length of ischemia, the organ which is affected, oxidation-reduction reactions in the environment, existence of cofactors and the time of onset of reperfusion. When a high concentration of NO is present in an oxidative environment, peroxynitrite will be produced, which worsens the situation. There is strong agreement that NO produces distinct and different effects depending on its concentration reached at the ischemic site [26].
It is shown that NO plays a key role in triggering the ischemic conditioning phenomenon in various organs via its antioxidant, anti-apoptotic and anti-inflammatory properties [27]. Nitric oxide production by eNOS may be involved in remote IPC and POC protection in the post-ischemic organs such as the heart and the kidneys. In a cardiac model of IR, Tang et al. demonstrated that remote POC inhibits the reduction of eNOS expression [28]. Studies by Yamasowa et al. [29] and Mahfoudh-Boussaid et al. [30] also demonstrated that eNOS expression is increased in the post-ischemic rat kidney with IPC treatment. Furthermore, Liu et al. concluded that the protective effect of post-conditioning in the kidney is closely related to the NO production following the increase in eNOS and iNOS expression [31]. These studies are in line with our results with the RPEC method in which the eNOS expression in the kidney of RPEC-treated animals was significantly higher compared to sham and IR groups.
Inducible NOS can be synthesized by many cell types in response to different cytokines and during stress conditions such as IR. Normal vascular function is closely related to this enzyme, which produces huge amounts of NO as a defense mechanism [32]. In the present study, the IR group showed significantly higher iNOS expression compared with the sham group. Interestingly, iNOS expression was also significantly higher in the RPEC group compared with the non-treated IR group. Moreover, 1400W worsens functional and morphological conditions in the RPEC-treated animals. This controversy may be explained by the fact that increased NO production by iNOS in the RPEC-treated animals occurs in the microenvironment in the absence of injurious molecules such as ROS and pro-inflammatory cytokines. However, in the IR group the overproduction of NO takes place in a situation which makes this molecule susceptible to produce reactive nitrogen species (RNS). In the renal IR model, the expression and activity of iNOS increase 24 h after local IPC [33]. It was shown that inhibition of iNOS prevents the protective effects of IPC in the kidney [34]. Thus, the elevated iNOS expression could be considered as an important component of protection induced by remote ischemic conditioning in the kidney.
In the present study, ameliorative effects of RPEC on the expression of pro-inflammatory cytokines and damaging molecules were abolished by L-NAME and 1400W, which suggests the important role of NO production by iNOS and eNOS. This is in line with previous studies demonstrating that NO down-regulates inflammatory reactions, which are important contributors to renal IR injury [35]. NO has been shown to mediate the expression of TNF-α, a well-known cytokine, which seems to induce the accumulation of neutrophils by indirect mechanisms involving the macrophages, and induces the production of other cytokines such as IL-6 [36]. NO also regulates neutrophil recruitment by inhibiting the expression of adhesion molecules in the vascular endothelium, resulting in increased blood flow to the ischemic tissues [37]. The effects of direct intrarenal administration of an NO donor on renal dysfunction and inflammatory reactions after IR were investigated by Rodriguez-Pena et al. [38]. They measured pro-inflammatory cytokines after blood flow re-establishment and found that treatment with the NO donor significantly lowered the pro-inflammatory cytokines. Furthermore, previous studies have demonstrated that remote ischemic conditioning provoke a systemic protective response involving modulation of immune cells either at post-translational level or through transcriptional regulation [39]. Moreover, in 2010 Yu et al. reported the protective effects of ischemic conditioning associated with decreasing levels of pro-inflammatory cytokines [40]. Altogether, it seems that the reduction of the inflammatory response by RPEC may be partly mediated through increased NO production.
Increased endogenous antioxidant system activity and reduced lipid peroxidation have been suggested as possible NO-dependent mechanisms against renal IR injury [41]. Our previous studies indicated that RPEC is associated with reduced oxidative stress in the renal tissue [15]. However, in the present study, the protective effect of RPEC on the oxidative state was abolished by L-NAME and 1400W. These effects suggest that NO production may have an important role in the attenuation of oxidative stress caused by renal IR.
In conclusion, the present study demonstrated a key role for the NO pathway in the protective effects of remote per-conditioning on the renal IR injury. The beneficial effects of RPEC on post-ischemic renal injury were abolished by pharmacological inhibition of NOS by L-NAME and 1400W, which was demonstrated by higher renal dysfunction, oxidative, inflammatory, and morphological damage. The protective effects of RPEC on renal IR injury may be closely related to the NO production after the reperfusion, and both eNOS and iNOS are involved in this protection.