Current issue
Archive
Manuscripts accepted
About the Journal
Editorial office
Editorial board
Section Editors
Abstracting and indexing
Subscription
Contact
Ethical standards and procedures
Most read articles
Instructions for authors
Article Processing Charge (APC)
Regulations of paying article processing charge (APC)
PULMONOLOGY / RESEARCH PAPER
Adenosine A2a Receptors Improve Hypoxic Pulmonary Arterial Hypertension Via Mitochondrial ATP-sensitive Potassium Channels
1
The First Affiliated Hospital of Wenzhou Medical University, China
Submission date: 2021-01-06
Final revision date: 2021-03-23
Acceptance date: 2021-05-03
Online publication date: 2021-05-03
Corresponding author
KEYWORDS
proliferationapoptosispulmonary vascular remodelingA2a receptormitochondrial ATP-sensitive potassium channels
TOPICS
ABSTRACT
Introduction:
This study is aimed to explore the effects of Adenosine A2a receptors (A2aR) on hypoxia-induced pulmonary hypertension (HPH) via mitochondrial ATP-sensitive potassium channels (MitoKATP) in vivo and in vitro.
Material and methods:
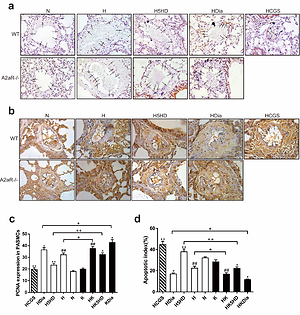
Using wild-type (WT) and A2aR-deficient (A2aR-/-) mice; hypoxic pulmonary artery smooth muscle cells (PASMCs) were induced by a 24-hours hypoxia exposure. Mice and PASMCs were treated with the A2aR agonist CGS21680, MitoKATP blocker 5-hydroxydecanoic acid sodium salt (5HD), or MitoKATP agonist diazoxide. Mitochondrial morphology was observed by electron microscopy. The mitochondrial membrane potential (Δψm); invasive hemodynamic parameters; right ventricular (RV) hypertrophy index; pulmonary arterial remodeling index; proliferative and apoptotic indexes; protein expression levels of A2aR, Bax, Bcl-2, and Caspase-9; and release of cytochrome C from the mitochondria to the cytoplasm were measured.
Results:
In vitro, hypoxia induced the opening of MitoKATP. The up-regulation of A2aR reduced the opening of MitoKATP, and the blocking of MitoKATP or activating A2aR promoted mitochondria-dependent apoptosis of PASMCs. In vivo, compared with WT mice, A2aR-/- mice displayed increased RV systolic pressure, RV hypertrophy index, and pulmonary arterial remodeling index. The expression levels of Bax, cytochrome C, and Caspase-9 were higher and Bcl-2 expression was lower in A2aR-/- mice than in WT mice. CGS21680 could reverse hypoxia-induced hemodynamic changes, RV hypertrophy, and pulmonary arterial remodeling as well as abnormal proliferation and apoptosis resistance in WT mice with pulmonary hypertension (PH).
Conclusions:
A2aR induced the mitochondrial-dependent apoptosis pathway and inhibited PASMC proliferation by blocking MitoKATP, thereby inhibiting pulmonary vascular structural remodeling and reducing PH.
This study is aimed to explore the effects of Adenosine A2a receptors (A2aR) on hypoxia-induced pulmonary hypertension (HPH) via mitochondrial ATP-sensitive potassium channels (MitoKATP) in vivo and in vitro.
Material and methods:
Using wild-type (WT) and A2aR-deficient (A2aR-/-) mice; hypoxic pulmonary artery smooth muscle cells (PASMCs) were induced by a 24-hours hypoxia exposure. Mice and PASMCs were treated with the A2aR agonist CGS21680, MitoKATP blocker 5-hydroxydecanoic acid sodium salt (5HD), or MitoKATP agonist diazoxide. Mitochondrial morphology was observed by electron microscopy. The mitochondrial membrane potential (Δψm); invasive hemodynamic parameters; right ventricular (RV) hypertrophy index; pulmonary arterial remodeling index; proliferative and apoptotic indexes; protein expression levels of A2aR, Bax, Bcl-2, and Caspase-9; and release of cytochrome C from the mitochondria to the cytoplasm were measured.
Results:
In vitro, hypoxia induced the opening of MitoKATP. The up-regulation of A2aR reduced the opening of MitoKATP, and the blocking of MitoKATP or activating A2aR promoted mitochondria-dependent apoptosis of PASMCs. In vivo, compared with WT mice, A2aR-/- mice displayed increased RV systolic pressure, RV hypertrophy index, and pulmonary arterial remodeling index. The expression levels of Bax, cytochrome C, and Caspase-9 were higher and Bcl-2 expression was lower in A2aR-/- mice than in WT mice. CGS21680 could reverse hypoxia-induced hemodynamic changes, RV hypertrophy, and pulmonary arterial remodeling as well as abnormal proliferation and apoptosis resistance in WT mice with pulmonary hypertension (PH).
Conclusions:
A2aR induced the mitochondrial-dependent apoptosis pathway and inhibited PASMC proliferation by blocking MitoKATP, thereby inhibiting pulmonary vascular structural remodeling and reducing PH.
Share
RELATED ARTICLE
| eISSN: | 1896-9151 |
| ISSN: | 1734-1922 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.