Introduction
Elevated plasma levels of low-density lipoprotein cholesterol (LDL-C) are a causal risk factor for atherosclerotic cardiovascular disease (ASCVD), which is the main cause of morbidity and mortality worldwide [1]. The major pathway of LDL-C clearance from the bloodstream is the uptake of LDL particles by hepatic LDL receptors (LDLRs) [2, 3]. Proprotein convertase subtilisin/kexin type-9 (PCSK9) is another critical regulator of circulating LDL-C levels that, following synthesis, predominantly in the liver, is secreted in plasma to act as a molecular chaperone to direct LDLR degradation [2, 4, 5]. PCSK9 binds to the extracellular domain of LDLR at the surface of hepatocytes, forming a complex that is endocytosed and transported to lysosomes for degradation. This process inhibits LDLR recycling and results in increased levels of LDL-C in plasma [6]. Therefore, increasing LDLR activity and inhibition of PCSK9 are effective therapeutic approaches to reduce circulating LDL-C levels and prevent the development of ASCVD [7–9]. Although most of the research has focused on the interaction between secreted PCSK9 and cell-surface LDLR, it is likely that intracellular PCSK9 also contributes to LDLR degradation [10]. Moreover, the mechanism of action of PCSK9 is not limited to LDLR degradation and involves the regulation of receptors of macrophages with a key role in the loading of lipids, formation of foam cells and atherosclerotic plaques, plaque stability, and vascular inflammation [11]. In this regard, various interventional studies have shown that using lipid-lowering drugs such as statins, despite a significant reduction in LDL-C levels through the increase of the LDLR, is less effective in reaching LDL-C targets in a considerable number of patients [12]. Moreover, statin intolerance due to side effects such as the development of hepatic damage, myalgia, or polyneuropathy is another factor necessitating alternative LDL-lowering therapies [13, 14]. In addition, while approaches utilized for reducing the plasma level of PCSK9 or inhibiting LDLR binding through monoclonal antibodies and antisense oligonucleotides are available, small molecule inhibitors are still being actively sought owing to their lower costs and ease of administration [15]. One such small molecule inhibitor is NYX-PCSK9i, which is safe and well tolerated in mice while exerting a potent lipid-lowering effect mainly on the non-HDL fraction [16]. However, finding new effective and safe lipid-lowering agents is still necessary to prevent ASCVD. Recently, several studies have suggested a beneficial role for functional foods, natural products, nutraceuticals, and in particular dietary polyphenols in cardiometabolic diseases [17–21]. Their regulatory effect on lipid metabolism includes the entire process from inhibiting enteric lipid absorption to promoting hepatic excretion of cholesterol as follows: lipid absorption reduction through reducing the cell permeability of enteric cells, cholesterol, and triglyceride synthesis inhibition along with a reduction in fatty acid uptake, elevation of reverse cholesterol transport and catabolism, and finally modulation of intestinal flora [22]. As stated by the International Lipid Expert Panel (ILEP), it seems that nutraceutical therapies are safe and well-tolerated options for the management of dyslipidemia [23], and their use is recommended in people who are statin-intolerant [24].
Ellagic acid (EA) is an active natural polyphenol and a hydrolyzed form of ellagitannins, which is found in some fruits and nuts such as pomegranates and walnuts [25].
Under physiological conditions, this polyphenol is metabolized by the gut microbiota to produce different types of urolithins [25]. Hitherto, numerous pharmacological effects have been reported for EA including antibacterial, anti-inflammatory, preventive, and therapeutic effects on obesity, hepato-protective effects, and anti-tumoral effects [26, 27]. Antiatherogenic properties of EA have been previously reported in several experimental and animal studies. EA had a direct impact on lipid metabolism in vitro [28], and could also reduce the plasma levels of cholesterol in vivo [29]. In addition, it has been reported that EA could increase the expression of genes involved in cholesterol metabolism, such as LDLR, at both the mRNA and protein levels in HepG2 cells [30, 31] as well as in the liver of diabetic mice [32]. However, due to the inter-individual variability in polyphenol metabolism as well as low intestinal absorbance, EA’s effectiveness in humans remains controversial [33]. Nevertheless, its driven metabolites, urolithins A (UA) and B (UB) have more bioavailability than EA and have also exhibited antiatherosclerotic properties through the regulation of cholesterol metabolism pathways [34–36]. It has been found that UA and UB could decrease lipid plaque deposition and attenuate atherosclerotic lesions in animal models through activation of the class B scavenger receptor (SR-BI) and induction of the reverse cholesterol pathway [35, 37]. Therefore, according to the reported antiatherosclerosis effects of UA and UB and the importance of LDL particle clearance by liver cells to reduce ASCVD, this study aimed to evaluate the possible role of UA and UB on the mRNA expression levels of LDLR and PSCK9 genes, and also of the uptake of LDL particles in HepG2 cells.
Material and methods
Materials
The tested compounds were obtained from Xi’an Le Sen Biotechnology Co., Ltd. (Xi’an, China). Other materials, including DMSO (d2650, Sigma, USA), trypsin-EDTA (15090046, Gibco, USA), RPMI 1640 medium (Dena Zist, Iran), fetal bovine serum (FBS) (10082139, Gibco, USA), antibiotics (penicillin/streptomycin) (15070063, Gibco, USA), Chloroform (107024, Merck, USA), ethanol (100983, Merck, USA), BIOzol RNA Lysis buffer (BN-0011.33, Bonyakhteh, Tehran, Iran), a cDNA synthesis kit (YT4500, Yekta Tajhiz Azma, Iran), DNase/RNase free water (CH8161, Sinaclon, Iran), and a QPCR master mix kit (BN-0011.17.4, Tehran, Iran), were used for tests.
LDL uptake in HepG2 cells
The hepatoma cell line HepG2 was used as the most common in vitro model utilized for studying the cholesterol-lowering action of nutraceuticals. The LDL uptake was detected by the LDL Uptake Cell-Based Assay Kit (KA1327, Abnova, Taiwan), according to the manufacturer’s protocol. Briefly, the HepG2 cells (10,000 cells/well) were seeded in a 96-well plate in RPMI 1640 medium enriched with 10% FBS and 1% antibiotics at 37°C in a humidified 5% CO2 atmosphere for 2 days and then treated with curcumin (20 μM) [38], berberine (50 μM) [39, 40], urolithin A (20–160 μM) [41] or urolithin B (20–160 μM) [42] and incubated at 37°C with 5% CO2 for an additional 24 h. To check the time-dependent properties of urolithin A (80 and 160 μM) and urolithin B (80 and 160 μM), a parallel test was also carried out with 48 h incubation. After incubation, the cells were changed to a serum-free medium containing LDL-DyLight 550 (0.01%) (10 μg/ml) followed by a further 24 h incubation at 37°C. At the end of the LDL uptake incubation, the culture medium was aspirated and replaced with fresh culture medium or PBS, and the degrees of LDL uptake were assessed using fluorescent microscopy (IX53, Olympus) with filters capable of measuring excitation and emission wavelengths of 540 and 570 nm, respectively. Followed by this, immunofluorescence staining of LDLR was also performed. According to the kit’s protocol, the cells were fixed and stained for LDLR using the Rabbit Anti-LDL Receptor Primary Antibody and DyLight 488-conjugated secondary antibody, and the staining was examined using fluorescence microscopy (IX53, Olympus) with a filter capable of excitation and emission at 485 and 535 nm, respectively. All treatments were performed in triplicate.
LDLR and PCSK9 expression in HepG2. Cell culture
HepG2 cells were grown in the RPMI 1640 medium enriched with 10% FBS and 1% antibiotics at 37°C in a humidified 5% CO2 atmosphere. Once the confluency was achieved to 70%, the cells (100,000 cells/well) were seeded in a 24-well plate (SPL) in normal serum medium for 48 h, and then transferred to medium supplemented with curcumin (20 μM) [38], berberine (50 μM) [38–40], urolithin A (80 μM) [41] or urolithin B (80 μM) [42] and incubated at 37°C with 5% CO2 for a further 24 h. All treatments were performed in triplicate. Three wells were also considered as controls without any treatment. After incubation, the medium was removed and cells were prepared for RNA extraction.
RNA extraction and cDNA synthesis
The BIOzol RNA Lysis buffer (BN-0011.33, Bonyakhteh, Tehran, Iran) was used to extract total cellular RNA. About 1 μg of total RNA with an absorbance of 1.8–2 at 260/280 nm (measured by a NanoDrop 2000 spectrophotometer (Thermo, Wilmington, DE, USA)) was used for the 1st strand cDNA synthesis using a cDNA synthesis kit (YT4500, Yekta Tajhiz Azma, Iran). cDNA synthesis was completed via the thermocycler device for 60 min at 37°C and 5 min at 70°C. The synthesized cDNA was stored at –20°C for future quantitative real-time PCR (qRT-PCR)
qRT-PCR
To evaluate the relative cell expression levels of LDLR and PCSK9 the SYBR Green qPCR method was applied (LightCycler 96, Roche Diagnostics, Mannheim, Germany) using the BON QPCR master mix kit (BN-0011.17.4, Tehran, Iran). The sequences of primers are shown in Table I. All reactions were carried out in duplicate. The Ct (cycle threshold) values of LDLR and PCSK9 were normalized to β-actin (ACTB) as an internal control, in each sample, and the comparative (2-ΔΔCt) method (fold change (FC)) was used to calculate the mRNA expression.
Table I
Sequences of primers used to evaluate the expression of LDLR and PCSK9
Statistical analysis
In vitro experiments were repeated three times and representative images are shown. The relative expression software tool (REST) was employed to compare the mRNAs’ fold changes of expression between the study groups. Data are presented as mean ± SE, and p-values of less than 0.05 were considered significant.
Results
LDL uptake
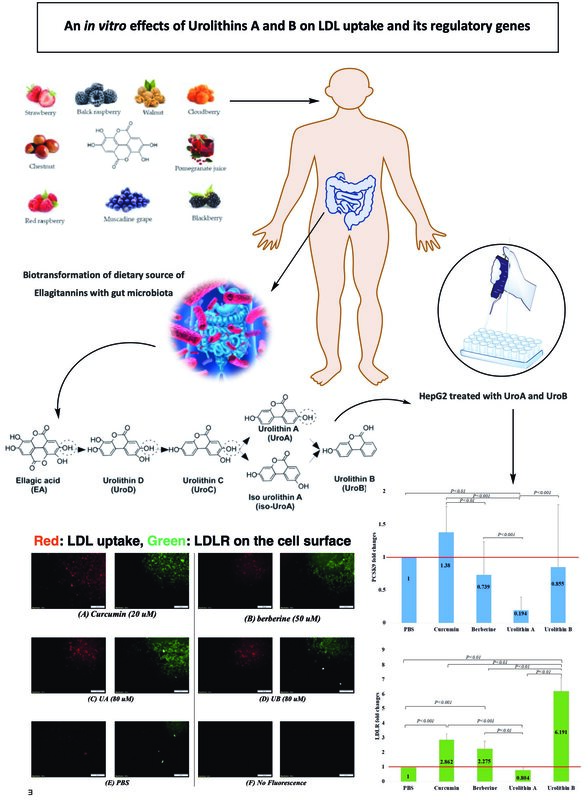
The results showed that 24-hour treatment of HepG2 cells with UA (20–160 μM) (Figure 1 A.1) and UB (20–160 μM) (Figure 1 B.1) increased LDL uptake as well as LDLR on the cell surface in a dose-dependent manner. However, after 48 h, both LDL uptake and cell surface of LDLR were reduced even at high concentrations (80 and 160 μM) of UA (Figure 1 A.2) and UB (Figure 1 B.2). So, the concentration of 80 μM and incubation time of 24 h were selected for the test.
Figure 1
LDL uptake (red) as well as LDLR on the cell surface (green) of HepG2 treated with (A) UA (20–160 μM), and (B) UB (20–160 μM) increase dose-dependently. However, they decreased after 48 h. The results shown are representative of an experiment repeated in triplicate for each treatment
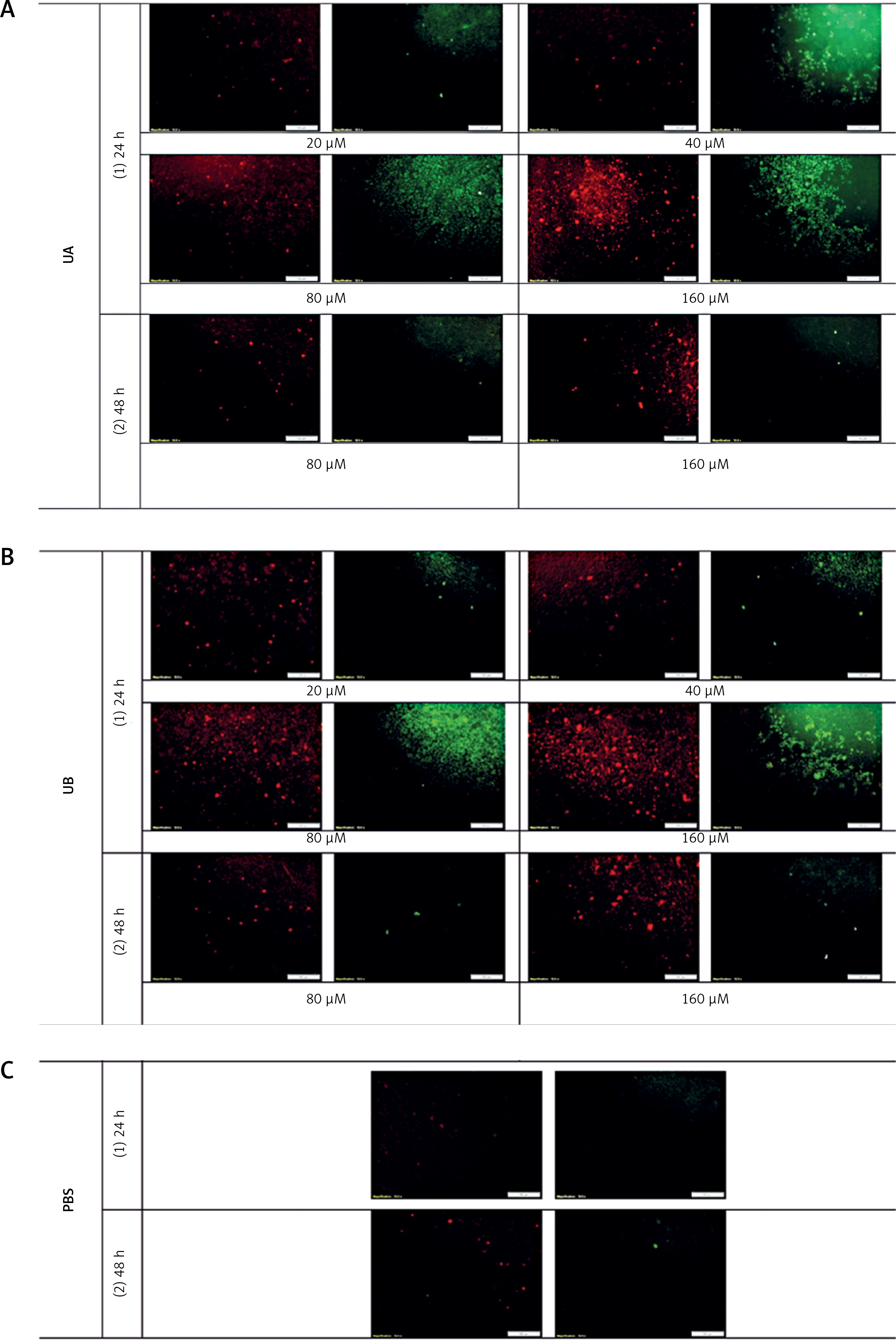
As shown in Figure 2, all treatments illustrated higher LDL uptake and cell surface LDLR when compared to the control (PBS treatment) (Figure 2 E). However, the LDL uptake and cell surface LDLR were higher in cells treated with UA (Figure 2 C) in comparison with cells treated with UB (Figure 2 D) and even in relation to the cells treated with curcumin (Figure 2 A) and berberine (Figure 2 B) as positive controls. However, UB, despite a considerable increase in LDL uptake and cell surface LDLR, showed no differences compared with curcumin and berberine treatments.
Figure 2
LDL uptake (red) as well as LDLR on the cell surface (green) of HepG2 treated with (A) curcumin (20 μM), (B) berberine (50 μM), (C) UA (80 μM), (D) UB (80 μM) after 24 h. (E) PBS and (F) No fluorescence. The results shown are representative of an experiment repeated in triplicate for each treatment
LDLR and PCSK9 expression
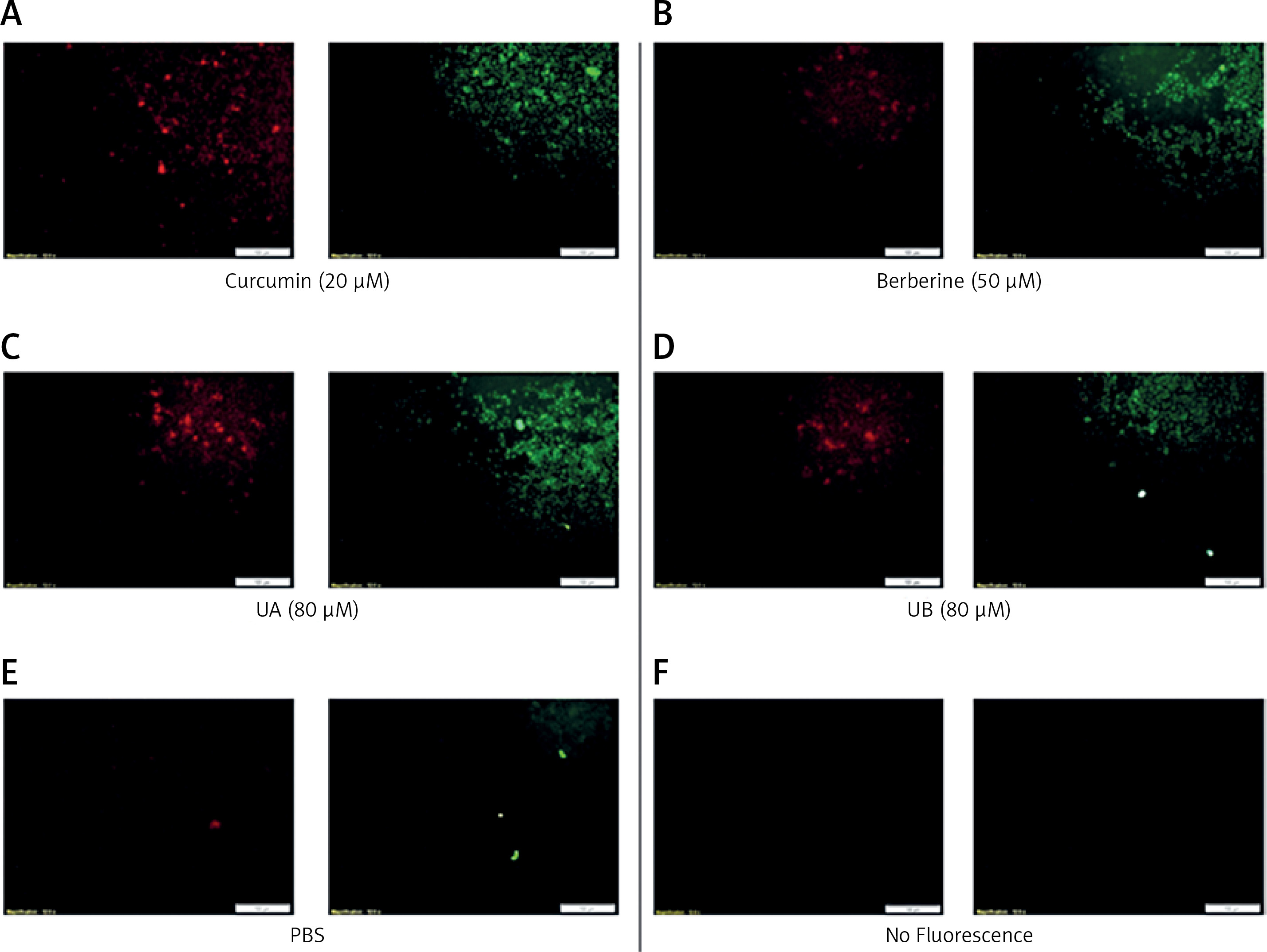
The results showed that all treatments except UA significantly increased the LDL expression levels of the cell surface of HepG2 (Figure 3 A) (at least more than 200%) in relation to the control (PBS treatment). Interestingly, UB indicated approximately 2 times greater LDLR expression levels in comparison with curcumin (FC = 2.144, p = 0.013) and berberine (FC = 2.761, p = 0.006) as positive controls. However, UA treatment resulted in 20% lower expression levels of LDLR on the cell surface of HepG2 in comparison with curcumin (FC = 0.274, p < 0.001) and berberine (FC = 0.352, p = 0.009) treatments. Moreover, UB demonstrated approximately 8 times higher LDLR expression levels when compared to UA (FC = 7.835, p = 0.001).
Figure 3
HepG2 LDLR (A) and PCSK9 (B) fold change expression in treatment groups. All data were normalized to the control group (PBS)
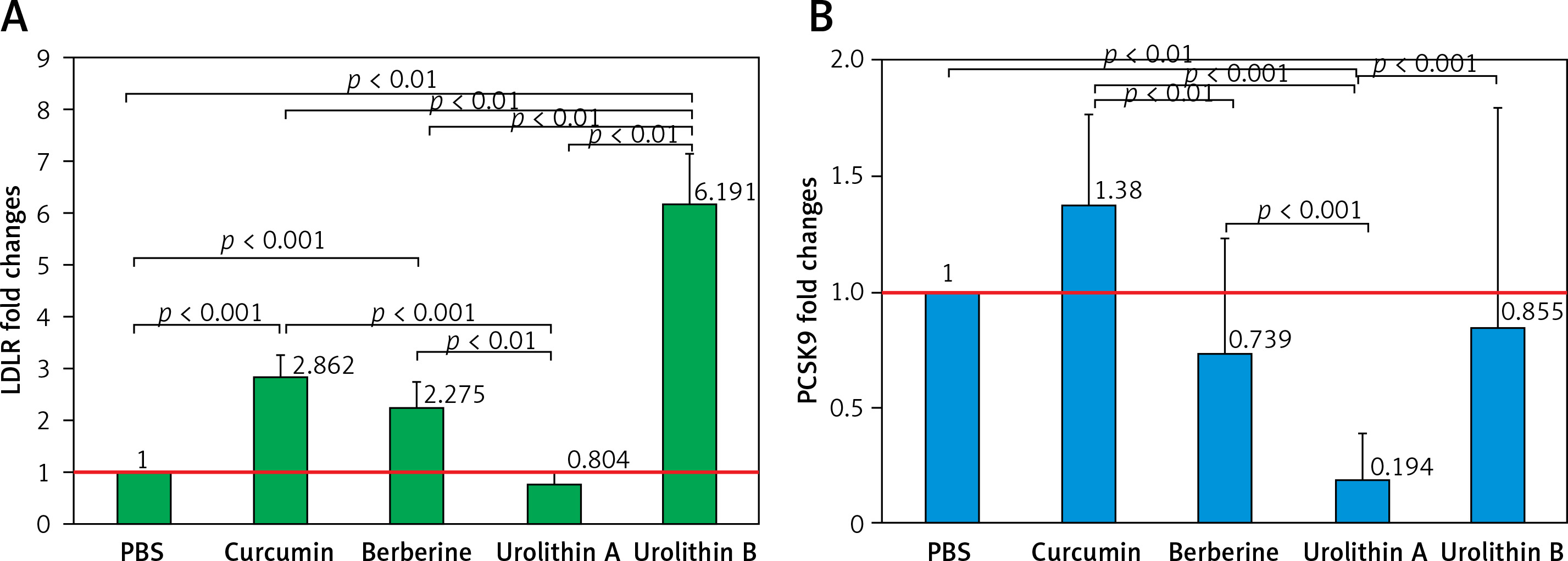
In contrast, PCSK9 showed lower expression levels in HepG2 cells treated with berberine, UA, and UB in relation to the control group (PBS treatment), though the difference was significant only for UA (FC = 0.194, p < 0.001). Also, berberine showed significantly lower PCSK9 expression levels compared with curcumin treatment (FC = 0.548, p = 0.035). Moreover, in comparison with UB as well as curcumin (FC = 0.140, p < 0.001) and berberine (FC = 0.255, p < 0.001) as positive controls, UA was more efficient in reducing PCSK9 expression levels in HepG2 cells. Although UB did not show any significant differences compared with curcumin and berberine treatment, it showed higher levels of PCSK9 expression when compared to the UA group (FC = 3.694, p < 0.001).
Discussion
There has been a recent trend in the use of natural products such as polyphenols to improve lipid profile and decelerate ASCVD [43, 44]. Natural products modulate lipid metabolism through a variety of mechanisms including anti-oxidative stress, anti-inflammation, decreasing the uptake and trafficking of fatty acid in hepatocytes, hepatic de novo lipogenesis reduction, enhancing lipolysis, induction of lipophagy, and increasing fatty acid β-oxidation [45].
The results of the present study showed notably higher LDL uptake and cell-surface LDLR in HepG2 cells treated with curcumin and berberine as two polyphenols with documented positive roles in cholesterol metabolism and enhancement of LDL uptake pathways. Additionally, this study demonstrated that both UA and UB are effective in promoting LDL uptake as well as in increasing the availability of cell-surface LDLR in HepG2 cells, though UA had a greater effect compared with the other three tested compounds. It was previously shown that curcumin by suppressing PCSK9 expression in hepatic cells could increase cell surface LDLR density and LDLR activity [38]. However, in this study curcumin did not significantly decrease PCSK9 expression levels, though LDLR expression level was considerably increased following curcumin treatment, and this might be the reason for increased LDL uptake and cell surface LDLR, which was observed in the present study. It has also been demonstrated that curcumin could up-regulate LDLR expression via the sterol regulatory element binding protein (SREBP) pathway in HepG2 cells [46] as follows: when SREBP binds to the promoter of HMGCR, whose expression or activity regulation has a critical role in cholesterol biosynthesis, the cholesterol biosynthesis speeds up. SREBP could also bind to the promoter of LDLR that is responsible for hepatic clearance of LDL-C. An increased level of cellular cholesterol acts as a negative controller of HMGCR expression [47]. SREBP-2 is a simultaneous activator of both LDLR and PCSK9 expression [48]. Berberine, as another effective cholesterol-lowering phytochemical [49], has been shown to increase LDLR expression in vivo and promote LDL uptake in vitro [50]. The results of the present study also illustrated increased in vitro LDL uptake following berberine treatment, which might occur due to increased LDLR expression levels. Additionally, although berberine exerted anti-PCSK9 activity in some previous studies [51, 52], this study failed to show decreased expression levels of PCSK9 in HepG2 cells after berberine treatment. Nevertheless, in comparison with curcumin treatment, the HepG2 PCSK9 expression level was significantly lower in cells treated with berberine. Moreover, another study reported that berberine through promoting foam cell formation by inducing scavenger receptor A (SR-A) expression in macrophages could counter-balance the beneficial effect of lowering serum cholesterol and subsequently lead to the development of atherosclerosis [53].
Recently, various clues to the role of gut microbiota and its related metabolites in atherosclerosis development and progression have emerged [54]. Urolithins are secondary metabolites of the gut microbiome transformation of the natural polyphenols ellagitannins and ellagic acid [55], which are found in some dietary products including certain nuts and fruits. Urolithins are derived from dibenzopyran-6-one and their differences are in hydroxyl group substitutions [56]. UroM5, UroD, UroE, UroM6, UroC, UroM7, UroA, IsoUroA, UroB, and IsoUroB are step-by-step products of biotransformation of ellagic acid to intermediate and final products through hydrolysis, decarboxylation, and dihydroxylation reactions. The concentration of urolithins as a consequence of inter-individual differences in gut microbiota varies from person to person and can be categorized as metabotypes A, B, and 0: the first is UroA producers; the second is Uro-A, IsoUro-A, and Uro-B producers; and the last one is Uro-non-producers [57].
Urolithins are correlated with different cardiometabolic factors. While UroA is correlated with apolipoprotein A-I and intermediate-HDL cholesterol, UroB and IsoUroA are correlated with LDL, VLDL, total cholesterol, oxidized-LDL, apolipoprotein B:apolipoprotein A-I ratio, and apolipoprotein B, and even could be potentially used as biomarkers of cardiometabolic disease [58].
Although the beneficial protective effect of UroB against cardiovascular disorders is mediated mainly through increasing the efflux of cholesterol from macrophages, the safety of UroB for clinical studies should be investigated. The other product of ellagic acid biotransformation, UroA, has supporting evidence of anti-atherosclerotic activity, which is partly related to its mitophagy-inducing activity. UroA also has an acceptable safety profile that facilitates its use in clinical trials [57].
This study for the first time evaluated in vitro effects of UA and UB, as microbiota metabolites with antiatherosclerotic properties [34–36, 59], on LDL uptake and its related genes. Interestingly, the results of the present study indicated that UA was more efficient in promoting LDL uptake as well as cell surface LDLR in HepG2 cells compared with the other treatments. Although the LDLR expression showed lower levels in the UA group compared with the other tested compounds, the significantly greater reduction of PCSK9 expression levels might be the reason for increasing the availability of LDLR on the cell surface and subsequent LDL uptake in HepG2 cells treated with UA. In contrast, UB was found to have a strong impact on the expression of LDLR in HepG2 cells, but it did not significantly reduce the expression level of PCSK9. To the best of our knowledge, there has been no previous document on the possible role of UA and UB in regulating LDL uptake, while the antiatherosclerotic properties of these compounds were previously suggested [60–62]. It was shown that UA could reduce ox-LDL-induced cholesterol accumulation in macrophages and promote cholesterol efflux from cells, hence interfering with cholesterol metabolism possibly through the reverse cholesterol transport pathway [61]. Similarly, UB was also reported to be able to induce reverse cholesterol transport via modulation of the expression of SR-BI and ATP-binding cassette (ABC) transporter A1 (ABCA1) and reverse lipid uptake by macrophages. These mechanisms provide a basis for the application of urolithins as potential antiatherosclerotic compounds [62]. In addition, it was demonstrated that UA through inhibition of lipase, α-glucosidase and dipeptidyl peptidase-4 could prevent metabolic-associated disorders such as diabetes and its related complications [63]. Nevertheless, some experiments have revealed that once urolithin is absorbed into the blood circulation, it conjugates mainly with glucuronide and sulfate, and these metabolites have lower biological activity compared with urolithin alone, so this is one of the limitations of this study, and the formulation and targeted delivery of urolithins is an important task for exploiting the therapeutic potential of these compounds [64]. There have been recent efforts in this direction, and a PEGylated liposomal formulation of UA was prepared and characterized, and showed acceptable morphological properties, size distribution, and sustained release profile [65]. Another area worthy of investigation which was not considered in this study would be a detailed assessment of LDLR-PCSK9 interaction in response to urolithin treatment. Further evidence from clinical studies would also be necessary to confirm the LDL-lowering activity of urolithins, particularly UA and UB, in clinical practice. Such clinical trials would be more feasible for UA since the safety of this compound has already been confirmed in clinical studies [66, 67]. The importance of in vivo studies to confirm the beneficial effects of UA relates mainly to the fact that in many in vitro studies there are concerns about the non-physiological concentrations used. Moreover, in vitro studies usually ignore the role of the two key factors of metabolism and tissue distribution. Therefore, confirmation of the present findings in in vivo models could provide robust evidence on the mechanism of the lipid-regulating action of urolithins. Some other possibilities for future studies could be using conjugated forms of urolithins and stratification of participants according to the metabotype responsiveness of their gut microbiota ecology [68]. Finally, it remains to be determined whether the anti-atherosclerotic action of urolithins can also be attributed to lipid-independent actions of these compounds such as anti-inflammatory and antioxidant activities, and whether these compounds can exert clinically relevant pleiotropic effects like what is known and well established for statins [69–72].
In conclusion, the present results suggest that UA was more effective than UB in promoting LDL uptake as well as cell surface LDLR in HepG2 cells. This activity seems to be mostly mediated through the suppression of PCSK9 expression but not the induction of LDLR expression. These findings support the promising potential of UA for the treatment of atherosclerosis, which needs to be confirmed in future in vivo studies. Also, mechanistic investigation of the antiatherosclerotic properties of UA is recommended.