Introduction
Acute liver injury (ALI) has been recognized as one of the most common global health problems and can be caused by hepatitis B viral infection, drug abuse, alcoholism, toxins, and other factors [1]. Long-term liver damage can progress to end-stage liver diseases, such as cirrhosis, chronic liver failure, and hepatocellular carcinoma [2]. However, there is currently no clinically effective drug for treating liver damage because the molecular mechanisms are still unclear. In recent years, increasing evidence has demonstrated that the molecular mechanisms of ALI are associated with regulatory T cell differentiation, NLRP3 activation, and apoptosis [3–5]. NLRP3 inflammasome-driven inflammatory damage is involved in the pathogenesis of various liver diseases, such as drug-induced liver injury, alcoholic liver disease, non-alcoholic fatty liver disease, and liver cancer [6]. During the development of liver injury, the NLRP3 inflammasome signaling pathway is activated and promotes the secretion of pro-inflammatory cytokines and chemokines and the subsequent recruitment of neutrophils and immune cells [7]. However, the regulatory mechanisms of the NLRP3 inflammasome signaling pathway during the pathological process of ALI are poorly understood.
The C6orf120 gene encodes a secreted N-glycosylated protein with largely unknown functions. In our team’s previous work, we revealed that C6ORF120 protein has an immunoregulatory function and is mainly expressed in liver tissues. Therefore, we prepared C6orf120-knockout rats and revealed the regulatory role of C6orf120 in the pathogenesis of concanavalin A-induced autoimmune hepatitis [4]. There are many etiologies of liver injury, such as drugs, immunity and toxins. Drug-induced toxicity is the most common cause of ALI [8]. However, the role of C6orf120 in drug-induced ALI has not been thoroughly investigated. Here, we hypothesized that C6orf120 is involved in the pathological process of drug-induced ALI.
The abuse of traditional Chinese medicine (TCM) is the main cause of DILI [9]. For DILI, there is currently no specific treatment, and the fundamental reason for the poor efficacy of so-called liver-protecting drugs is that the mechanism is unclear. Carbon tetrachloride (CCl4) is a classical chemical hepatotoxin that is widely used to induce acute hepatitis damage with an activated inflammation response [10]. CCl4-induced hepatic injury closely imitates the pathological changes and molecular mechanisms observed in patients suffering from drug-induced ALI [11]. In this study, we established a CCl4-induced ALI model and explored the underlying mechanisms of C6orf120 involved therein.
Material and methods
Animals and models
Male wild-type (WT) Sprague Dawley rats (weighing 200–220 g, 6–8 weeks of age) were obtained from the Laboratory Animal Center of Health Science Center, Peking University. Male C6orf120-knockout (C6orf120–/–) Sprague Dawley rats (weighing 200–220 g, 6–8 weeks of age) were supplied by Suzhou Saiye Biotechnology Co., Ltd. using TALEN-mediated gene knockout technology. The rats used in this study were fed in the Animal Center of Health Science Center, Peking University in a specific-pathogen-free environment. Five rats were housed in each cage and the temperature was controlled at 20–25°C at a humidity of 50 ±15%. Corncob padding, feed, and water were treated by high-temperature and high-pressure sterilization. All rats were observed for a week before the beginning of the experiment and randomly divided into four groups (n = 8 per group). WT and C6orf120-/- rats were intraperitoneally administered with a single dose of CCl4 (1 : 1 v/v in olive oil, 2 µl/g) for 24 h to establish CCl4-induced acute liver damage based on previously described protocols [12]. The control rats were injected with the same dose of olive oil. Rats were euthanized 24 h after CCl4 injection and liver tissues and blood samples were collected for analysis. All animal experiments complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals [13].
Hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC)
Rat liver tissues were dissected and fixed in 10% formalin. After dehydration and paraffin embedding, the liver tissues were cut into 4-µm sections and stained with H&E. The sections were imaged under a microscope (Zeiss AG, Oberkochen, Germany). The pathological grading of liver injury was evaluated by two independently trained authors (Mei-xin Gao and Xin-cheng Song) who were blinded to the group assignment.
For IHC analysis, rabbit anti-rat C6orf120 (Biorbyt) and anti-rat cleaved caspase-3 (Cell Signaling Technology) were used as primary antibodies. Biotinylated goat anti-rabbit IgG was used as the secondary antibody (ZSGB-BIO, Beijing, China). ImageJ software (NIH, Bethesda, MD, USA) was used to assess the expression of C6orf120 and cleaved caspase-3 by analyzing positive staining.
Alanine aminotransferase (ALT)/aspartate aminotransferase (AST) assessment
Serum samples were separated from blood by centrifugation at 2,000 rpm for 10 min at 4°C. ALT and AST levels in WT and C6orf120–/– rats were detected by a HITACHI apparatus according to the manufacturer’s protocols.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was isolated from rat liver with TRIzol reagent (TaKaRa Biotechnology Co., Ltd., Dalian, China) according to the manufacturer’s instructions. cDNA was synthesized using TransScript First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). RT-PCR was performed to analyze the expression levels of target genes using the PrimeScript RT reagent kit (TaKaRa, Shiga, Japan) according to standard protocols. The 7500 real-time PCR System (Applied Biosystems) was used to analyze the mRNA expression of glyceraldehyde 3-phosphate (GAPDH), interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α. GAPDH was amplified as a reference gene. The sequences of the primers are listed in Table I.
Table I
Primer sequences used in RT-PCR
Western blot
Liver tissues from WT and C6orf120–/– rats were immediately excised and mixed with tissue protein lysis buffer (Beyotime, Shanghai, China) containing 1 mM phenylmethane sulfonyl fluoride (Beyotime Biotechnology, ST506). After centrifugation, the proteins were denatured and the protein was loaded onto sodium dodecyl sulfate-polyacrylamide gels (10%) for electrophoretic separation and then transferred onto Immobilon polyvinylidene fluoride membranes (Sigma; EMD Millipore, Billerica, MA, USA; cat. no. P3313). After blocking with 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 (TBST), the membranes were incubated at 4°C overnight with primary antibodies against C6orf120, NLRP3, caspase 1, IL-1β, nuclear factor-κB, Bcl-2, and Bax (Abcam), as well as cleaved caspase-3, p-c-Jun N-terminal kinase (Cell Signaling Technology). The membranes were washed with TBST three times. Then, they were incubated with secondary antibodies (ZF-0316, ZSGB-BIO, Beijing, China) for 2 h at room temperature. The finally obtained immunoreactive protein bands were imaged using the Tanon Imaging system (Tanon Science and Technology Co., Ltd., Shanghai, China).
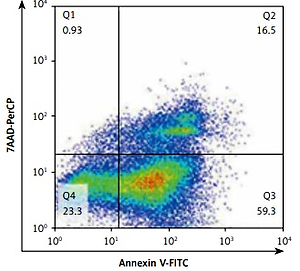
Induction of hepatocyte apoptosis and flow cytometry
Primary hepatocytes were isolated using a modification of the two-step collagenase perfusion method as mentioned previously [14]. The purification of hepatocytes was achieved via centrifugation over Percoll [15]. The hepatocytes in the experimental group were treated with medium containing 1% (v/v) CCl4 [16] and the control group was cultured in Dulbecco’s modified Eagle medium. The cells were plated in 6-well plates at a density of 5 × 106 cells/well and cultured under a humidified atmosphere of 5% CO2 at 37˚C for 24 h. After 24 h of CCl4 induction, the hepatocytes were harvested and Annexin V and 7-aminoactinomycin D (Biolegend) were added to stain apoptotic cells. The stained cells were quantified using the BD FACSCalibur flow cytometer.
Results
Expression of C6ORF120 in CCl4-induced ALI
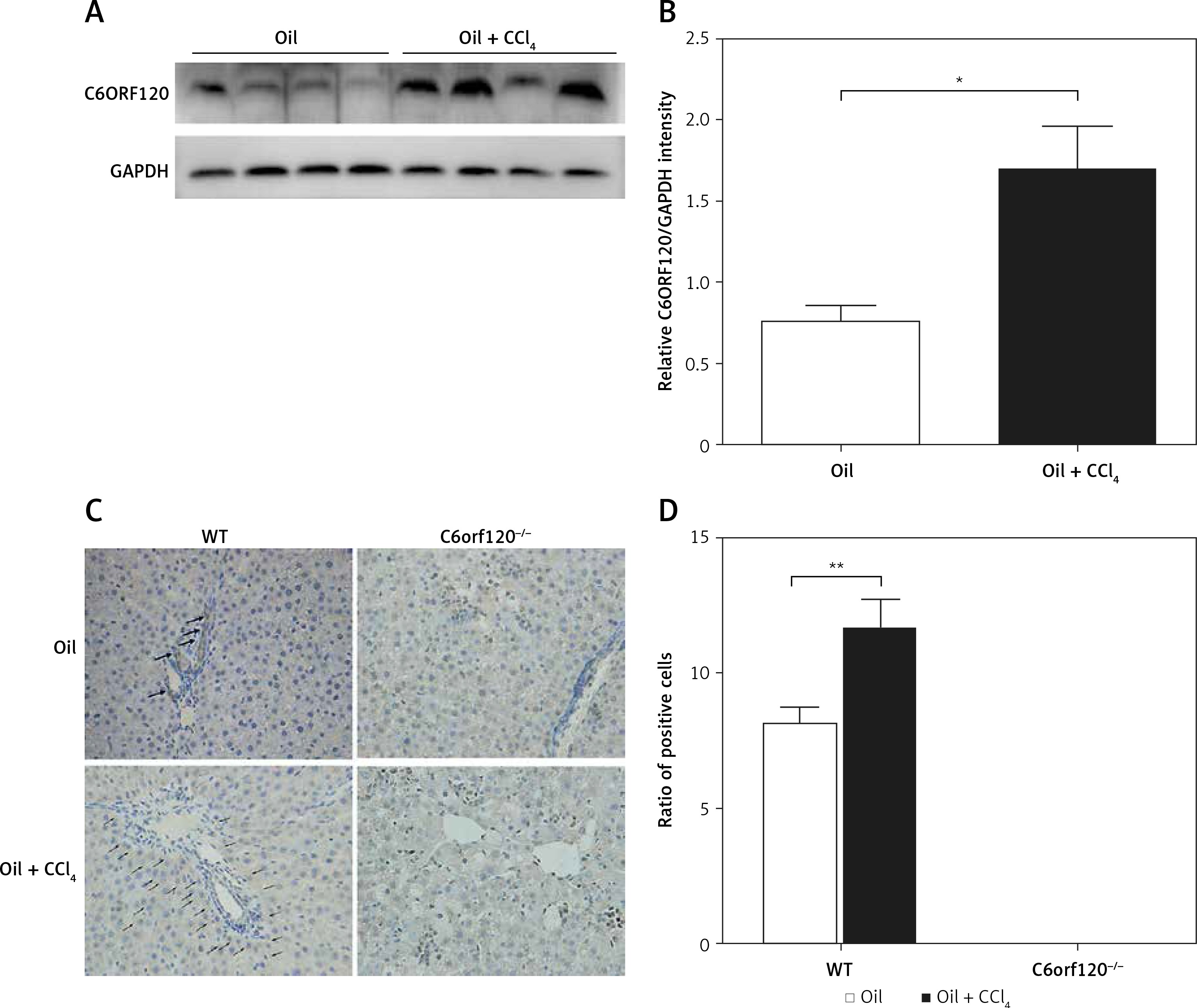
To investigate whether C6orf120 is involved in the development of ALI in rats, the protein expression of C6orf120 from rat liver tissues was determined by western blot 24 h after CCl4 injection. The results showed that C6ORF120 expression was significantly elevated in WT rats treated with CCl4 compared with that of the control group (p < 0.05) (Figures 1 A and B). Consistent with the results of western blot, IHC indicated that the ratio of C6orf120-positive cells in liver tissues of CCl4-treated rats was higher than that in the control group (p < 0.01) (Figures 1 C and D).
Liver injury was aggravated in C6orf120–/– rats
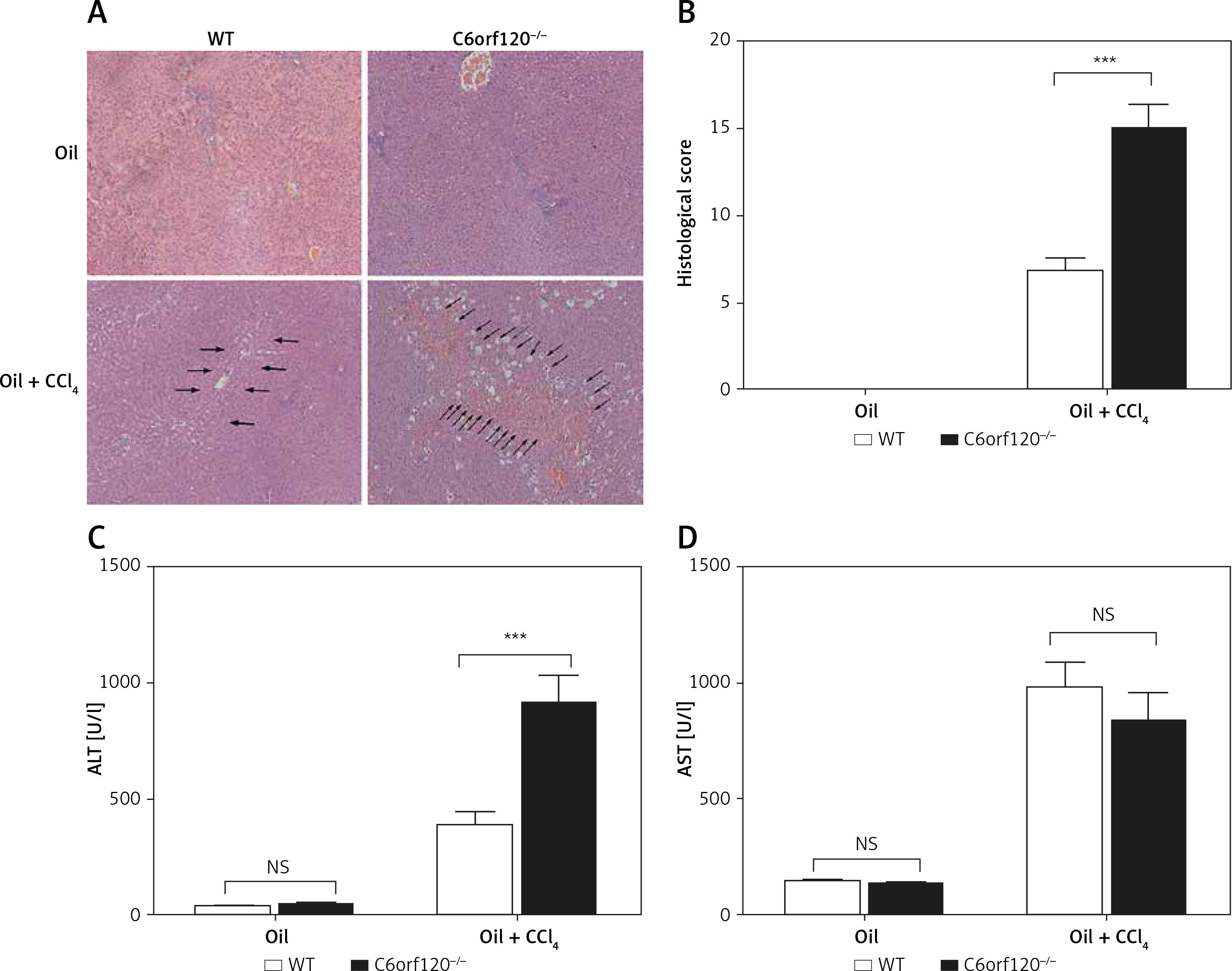
To evaluate the extent of liver damage, the pathological changes in CCl4-induced WT and C6orf120–/– rats were assessed by histopathology (Figures 2 A and B). In C6orf120–/– rats, H&E staining of liver tissues revealed severe pathological changes, as indicated by spotty or bridging necrosis, inflammatory cell infiltration, and ballooning degeneration compared with CCl4-treated WT rats (p < 0.001). Moreover, we tested the liver function of WT and C6orf12–/– rats 24 h after CCl4 injection. Both types of rats displayed hepatic dysfunction, as evidenced by increased ALT and AST levels (Figures 2 C and D). The level of ALT was much higher in C6orf120–/– rats (WT: 388.7 ±55.96 vs. C6orf120–/–: 915.9 ±118.8, p < 0.0001). Additionally, the levels of serum AST were increased in WT and C6orf120–/– rats after CCl4 administration, but there was no significant difference between the two groups (p > 0.05) (Figure 2 D).
mRNA levels of IL-1β, IL-6, and TNF-α were increased in liver tissues of C6orf120–/– rats
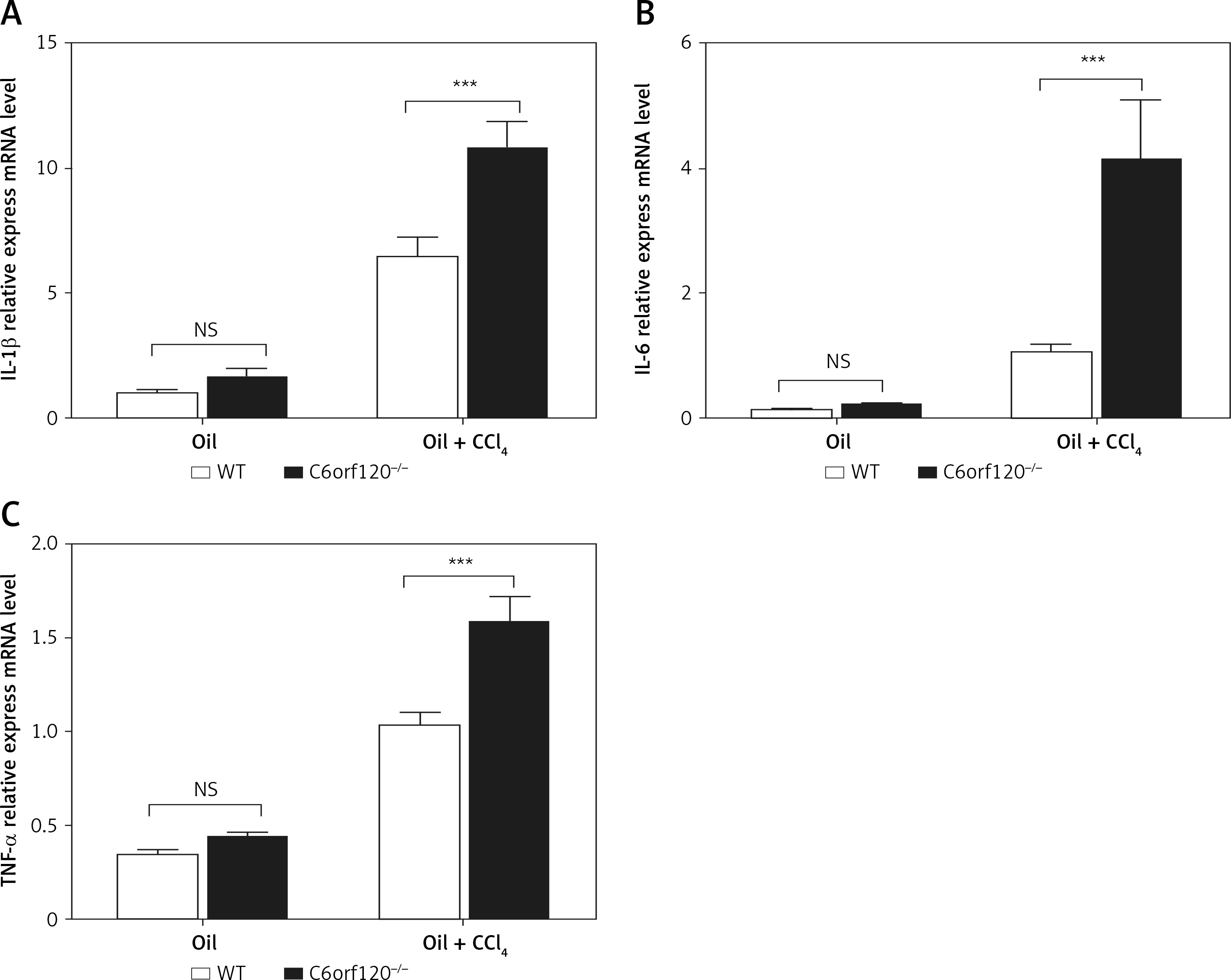
Hepatic injury induced by CCl4 is usually accompanied by an inflammatory reaction. To investigate whether C6orf120 is involved in the inflammatory response caused by CCl4, inflammatory cytokines in liver tissues were evaluated by qRT-PCR. The results showed that the expression levels of IL-1β, IL-6, and TNF-α in liver tissues increased significantly in C6orf120–/– rats compared to those in WT rats (p < 0.001) (Figures 3 A–C).
Expression of NLRP3 inflammasome was increased in C6orf120–/– rats
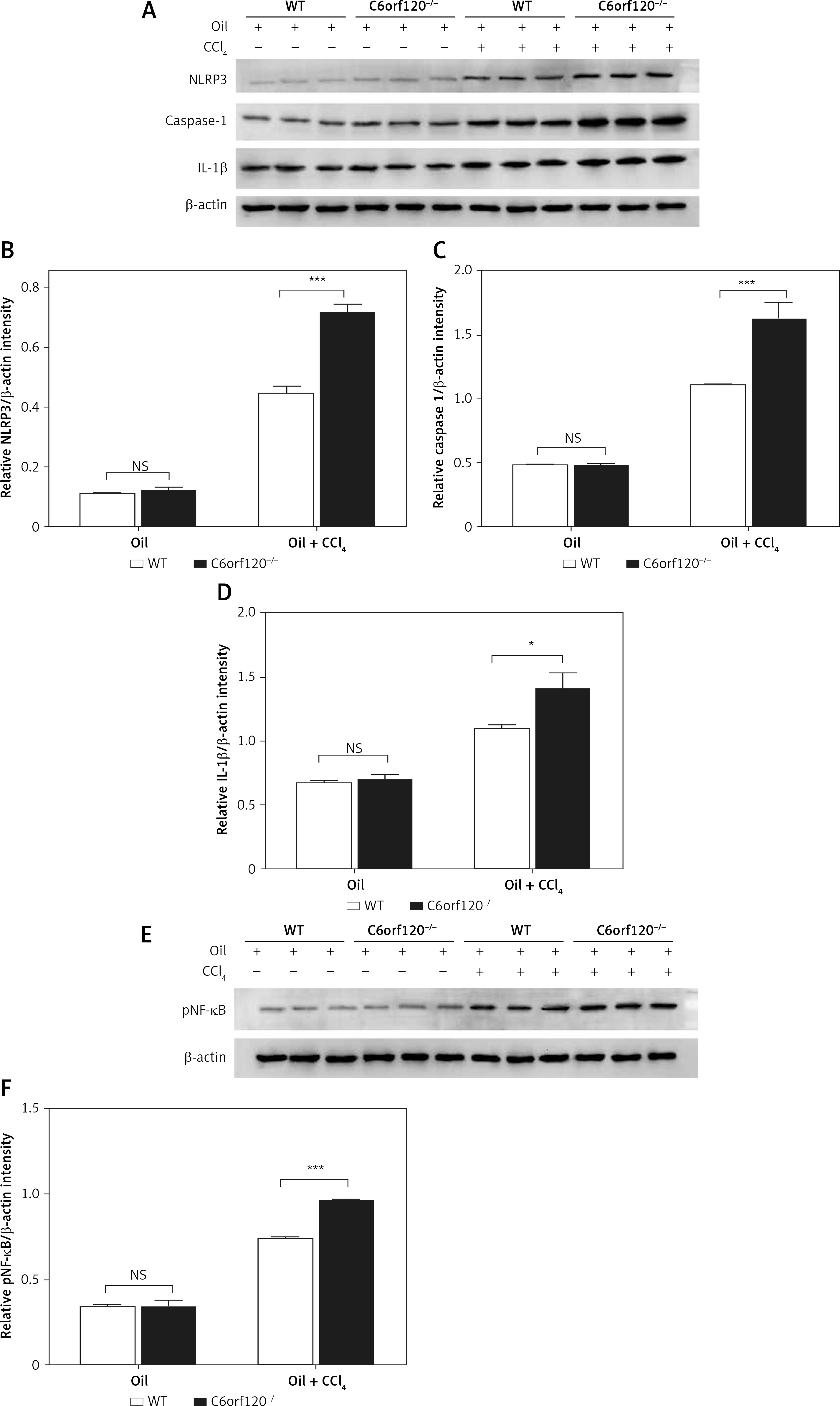
The NLRP3 inflammasome signaling pathway plays a crucial role in the modulation of liver inflammation [17]. To elucidate the possible molecular mechanisms of C6orf120 in CCl4-induced liver damage, the expression of NLRP3, caspase-1, and IL-1β in liver tissues was detected by western blot. The results showed that the expression levels of NLRP3, caspase-1, and IL-1β were distinctly upregulated in C6orf120-/- rat liver tissues compared with those in WT rats (p < 0.001, p < 0.001, p < 0.05, respectively) (Figures 4 A–D). Moreover, since NF-κB exerts essential functions in the inflammation process, we further evaluated the effect of C6orf120 on the expression of NF-κB. Western blot demonstrated that C6orf120 deficiency upregulated p-NF-κB expression in liver tissues (WT: 0.7380 ±0.01607 vs. C6orf120–/–: 0.9665 ±0.002297, p < 0.001) (Figures 4 E and F).
Figure 4
C6orf120 suppressed CCl4-induced hepatic injury by inhibiting NLRP3 inflammasome signaling pathway. A – Western blot of expression of NLRP3, caspase-1, and IL-1β in the liver. Relative density of protein bands for NLRP3 (B), caspase-1 (C), and IL-1β (D). E – Western blot of p-NF-κB expression in the liver. F – Relative density of p-NF-κB protein bands
C6orf120 deficiency exacerbated CCl4-induced hepatocellular apoptosis
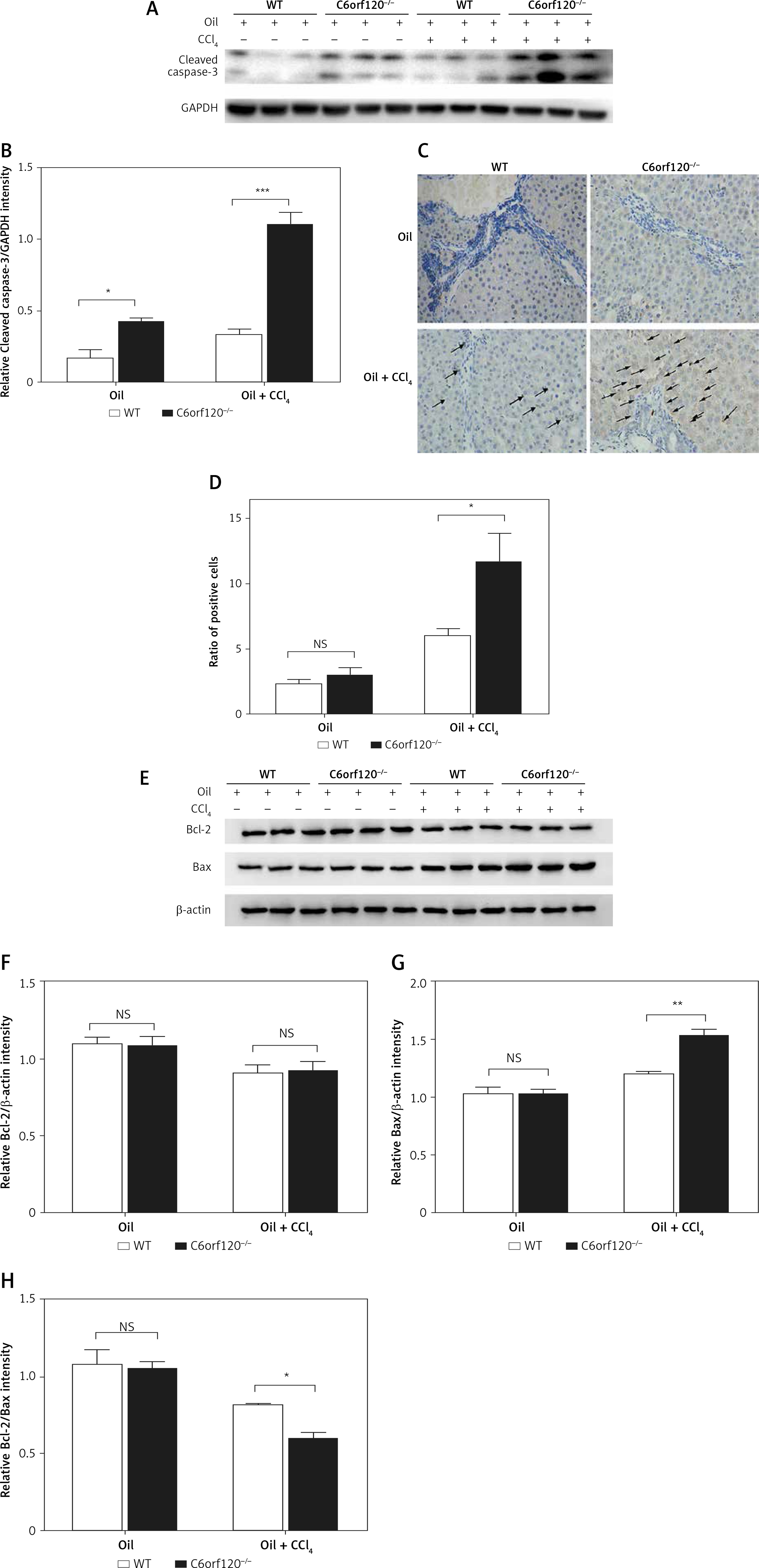
To evaluate the possible function of the C6orf120 gene on CCl4-induced apoptosis, the expression levels of cleaved caspase-3 were determined by western blot. The results showed that the expression of cleaved caspase-3 was significantly upregulated in C6orf120–/– rats compared to WT rats 24 h after CCl4 injection (WT: 0.3333 ±0.03779 vs. C6orf120–/–: 1.106 ±0.08313, p < 0.001). In the control group, the expression of cleaved caspase-3 was higher in C6orf120–/– rats than in WT rats (WT: 0.1693 ±0.05706 vs. C6orf120–/–: 0.4276 ±0.02145, p < 0.05) (Figures 5 A and B). IHC analysis of liver tissues showed a similar trend (p < 0.05) (Figures 5 C and D). To further clarify whether the C6orf120 gene is correlated with mitochondrial apoptosis, the protein levels of Bcl-2 and Bax were detected. The results showed that the expression of Bcl-2 was similar between WT and C6orf120–/– rats (p > 0.05) (Figures 5 E and F). However, the expression of Bax was apparently elevated in C6orf120–/–- rats compared to WT rats (WT: 1.200 ±0.02455 vs. C6orf120–/–: 1.532 ±0.05585, p < 0.01) (Figures 5 E and G), resulting in an imbalance in the Bcl-2/Bax ratio (p < 0.05) (Figure 5 H).
Figure 5
C6orf120 suppressed CCl4-induced hepatic injury by inhibiting hepatocellular apoptosis. A – Western blot of cleaved caspase-3 expression. B – Relative density of the cleaved caspase-3 protein bands. C – IHC of cleaved caspase-3 in liver tissues. D – Ratio of positive cells. E – Western blot of expression of Bcl-2 and Bax Relative density of protein bands for Bcl-2 (F), Bax (G), and Bcl-2/Bax (H)
Expression of JNK was increased in C6orf120–/– rats
JNK activation is tightly correlated with CCl4-induced mitochondrial dysfunction and hepatotoxicity. The expression level of p-JNK was detected to evaluate whether C6orf120 suppresses JNK activation against hepatocytic death in CCl4-induced liver injury. The results showed that the p-JNK expression was significantly increased in C6orf120–/– rats compared with WT rats (WT: 1.098 ±0.01627 vs. C6orf120–/–: 1.517 ±0.06239, p < 0.001) (Figures 6 A and B). Additionally, compared with JNK1, JNK2 appeared to be markedly activated.
Figure 6
C6orf120 suppressed CCl4-induced hepatic injury by inhibiting JNK signaling pathways. A – Western blot of p-JNK expression. B – Relative density of p-JNK protein bands C – Representative images of the gating strategy for necrotic/apoptotic cells among hepatocytes in vitro D – Frequency of necrotic/apoptotic cells among hepatocytes in vitro
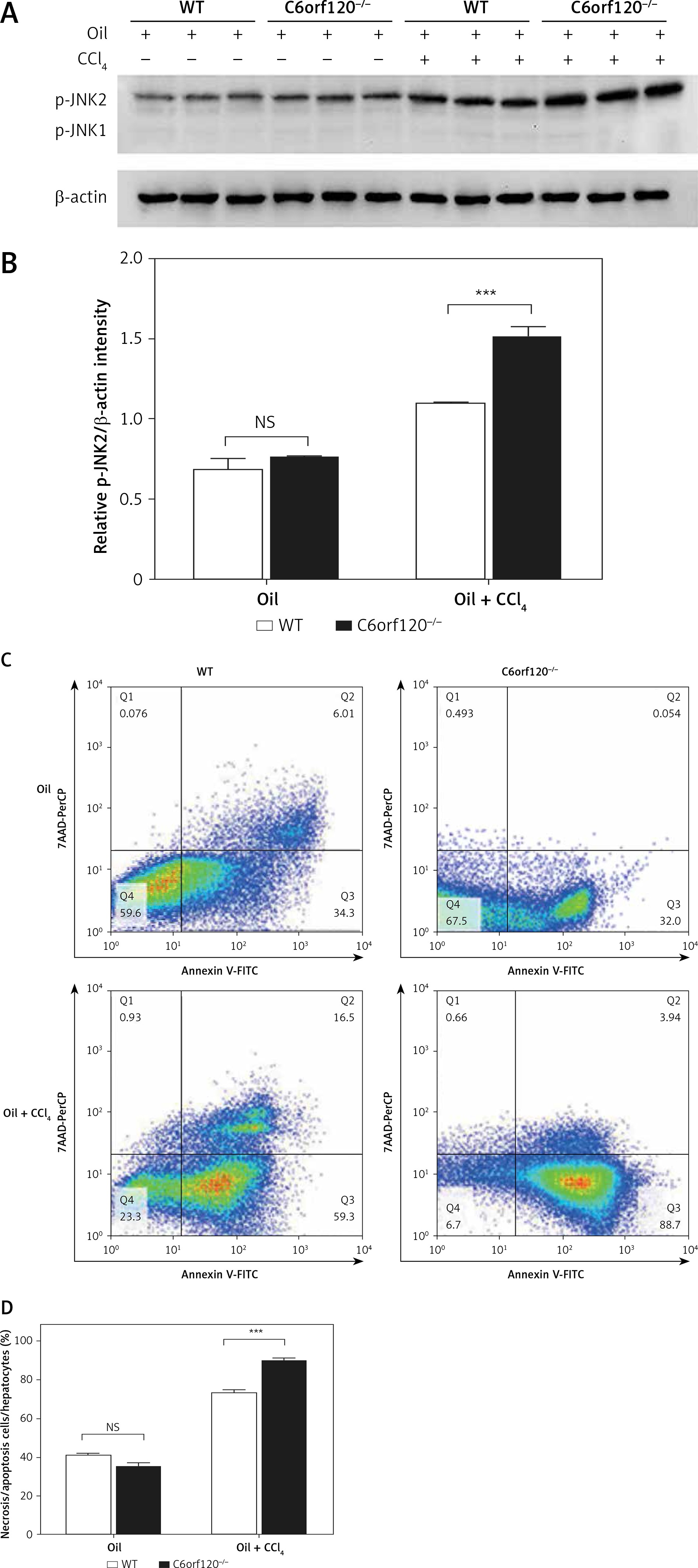
To further clarify the role of C6orf120 deficiency in hepatocellular apoptosis in vivo, primary hepatocytes from WT and C6orf120–/– rats were isolated and cocultured with CCl4 (1% v/v) for 24 h. Hepatocellular apoptosis analysis by flow cytometry showed that the frequency of apoptosis was significantly increased in hepatocytes derived from C6orf120–/– rats compared with hepatocytes from WT rats (p < 0.001) (Figures 6 C and D).
Discussion
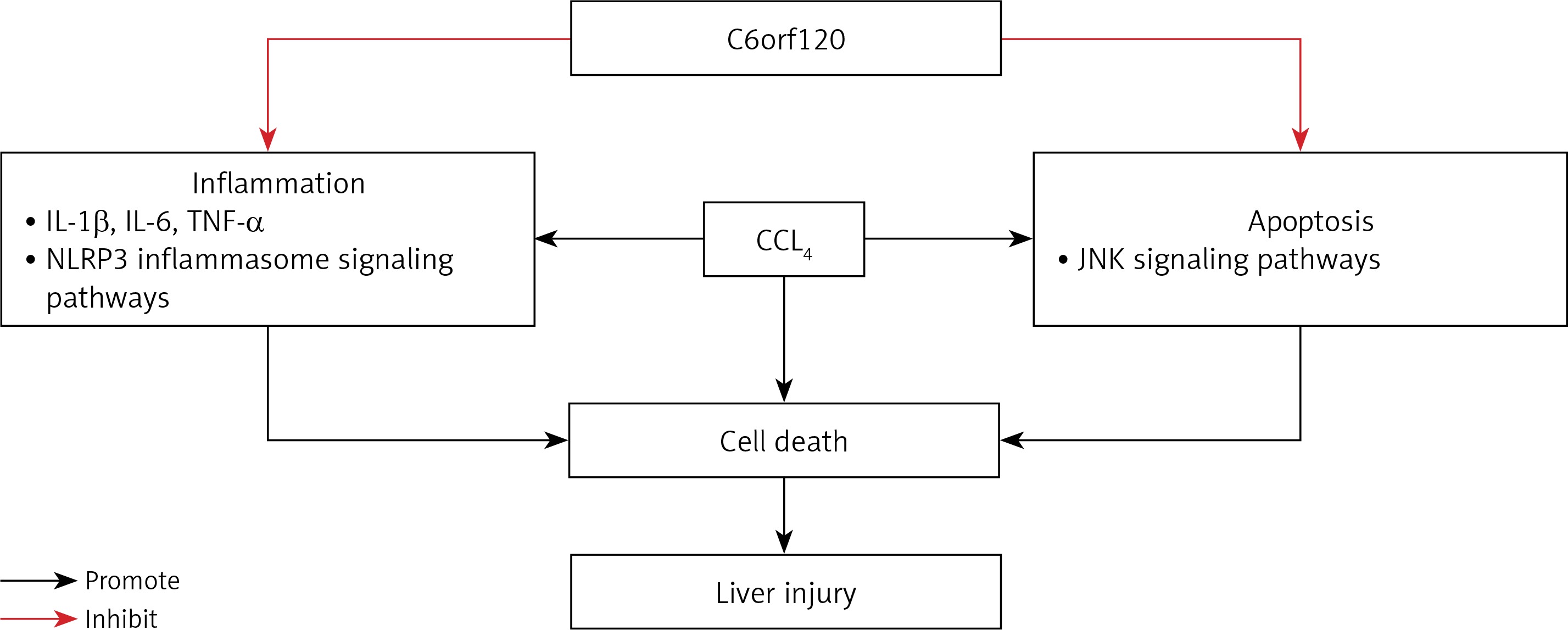
In this study, we revealed for the first time that C6orf120–/– rats were more susceptible to acute hepatic damage compared with WT rats, and the possible molecular mechanism of C6orf120 against CCl4-induced ALI might be related to the suppression of inflammation and apoptosis (Figure 7).
Carbon tetrachloride, a common hepatotoxin, has been widely applied to establish classical experimental liver injury models [18]. C6orf120–/– and WT rats were treated with CCl4 for 24 h to induce ALI, resulting in markedly increased ALT levels compared with those in WT rats, whereas there was no difference in AST levels. ALT is located in the cytoplasm of hepatocytes, and AST is located in both the cytoplasm and mitochondria. When hepatocytes suffer from acute damage, enzymes are mainly secreted from the cytoplasm and in turn ALT increases. When hepatocytes suffer from sustained or severe injury, enzymes from both the cytoplasm and mitochondria are released, resulting in a significant increase in AST [19]. Histopathological analysis via H&E staining further showed more severe degeneration and necrosis of hepatocytes in C6orf120–/– rats compared with those in WT rats, confirming that C6orf120–/– rats were susceptible to CCl4-induced liver injury than WT rats.
CCl4-induced liver injury is closely associated with the inflammatory response [20]. CCl4 and its metabolites probably stimulate Kupffer cells, which can secrete pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α. The pro-inflammatory cascade causes hepatocyte damage via the activation of macrophages and infiltration of neutrophils. In this study, the mRNA levels of IL-1β, IL-6, and TNF-α in liver tissues were significantly increased in C6orf120–/– rats compared with those in WT rats, suggesting that C6orf120 exerted hepatoprotective functions against CCl4-induced ALI by attenuating the secretion of pro-inflammatory cytokines.
To study the anti-inflammatory mechanism of C6orf120, the expression of p-NF-κB was also determined. The NF-κB signaling pathway plays an essential role in the pathological process of CCl4-induced acute liver damage [21, 22] and in mediating the secretion of inflammatory cytotoxins that aggravate liver damage. Our results showed that C6orf120 deletion increased the expression of p-NF-κB in liver tissues. To further investigate the protective mechanism of C6orf120, the involvement of NLRP3 inflammasomes in liver injury was investigated. NLRP3 inflammasomes are multi-protein complexes composed of intracellular pattern recognition receptors and are associated with the maturation and secretion of pro-inflammatory cytokines [23]. During the pathological process of acute liver damage, Kupffer cells or resident macrophages are activated and trigger inflammatory responses through common signaling pathways including NLRP3 inflammasomes, caspase-1, and IL-1β [24, 25]. In this study, 24 h after CCl4 administration, the expression levels of NLRP3, caspase-1, and IL-1β were significantly increased in C6orf120–/– rats compared with those in WT rats. Furthermore, increased levels of cytokines such as IL-1β can activate the downstream NF-κB signaling pathway, which can upregulate the expression of NLRP3 inflammasomes and the secretion of pro-inflammatory cytokines that aggravate inflammatory injury [26]. Our findings indicated that C6orf120–/– rats may be vulnerable to CCl4-induced liver injury by regulating the NLRP3/caspase-1/IL-1β/p-NF-κB signaling pathways.
Apoptosis and necrosis are the two major routes of cell death. To the best of our knowledge, necrosis is the main form of cell death in CCl4-induced hepatic injury. Studies performed by Yang et al. [27] and Campo et al. [28] revealed that hepatocyte apoptosis was involved in CCl4-induced ALI. Members of the Bcl-2 family, including Bcl-2 and Bax, are predominant regulators of the process of mitochondrial apoptosis. Bax is one of the most essential pro-apoptotic proteins that induces apoptosis, while Bcl-2 is an anti-apoptotic protein that inhibits Bax-induced apoptosis [29]. Caspase-3 is considered to be the most important terminal cleavage enzyme in apoptosis and an important part of cytotoxic T cell killing mechanisms. Activated Bax can stimulate caspase-3 and ultimately lead to caspase-dependent apoptosis [30]. In this study, the expression of Bax was markedly increased in C6orf120–/– rats compared with that in WT rats, whereas the expression of Bcl-2 was similar between the two groups. The results indicated that C6orf120 may inhibit CCl4-induced liver injury by suppressing mitochondrial apoptotic pathways. Meanwhile, the expression of cleaved caspase-3 was obviously elevated in C6orf120–/– rats compared with WT rats. Taken together, these observations suggested that C6orf120 protects against CCl4-induced ALI by regulating the mitochondrial apoptosis-associated molecules Bcl-2 and Bax to suppress caspase-3-dependent apoptotic signaling.
To further explore the mechanism of C6orf120 in hepatocyte apoptosis, the JNK signaling pathway was investigated. Several studies have shown that JNK signaling is vital in CCl4-induced mitochondrial dysfunction and hepatocyte death [31, 32]. In the process of CCl4-induced liver injury, sustained activation of JNK further promotes the transcription and expression of downstream apoptosis-related target genes, which trigger apoptosis via the mitochondrial pathway. Furthermore, p-JNK can directly mediate mitochondrial pathways by inhibiting the expression of Bcl-2 and promoting the expression of Bax in the cytoplasm [33]. Dhanasekaran et al. [34] reported that sustained activation of JNK induced Bcl-2 family-mediated apoptosis. In this study, C6orf120 deficiency aggravated the activation of p-JNK and downstream signaling pathways, suggesting that C6orf120 could prevent CCl4-induced liver injury by inhibiting JNK signaling.
There were some limitations in our study. First, it was widely reported that drug-induced liver injury was partly associated with immunoreaction. While our study only involved molecular biology research, experiments associated with immunological mechanisms are currently under way. Secondly, because C6orf120 is mainly expressed in liver and spleen tissues, the protective function of C6orf120 in spleen tissues should be assessed. Thirdly, we did not conduct experiments to demonstrate whether the expression of NLRP3 is directly related to C6orf120. Fourthly, if a group of previous CCl4 treatment had been added, the conclusion would have been sound and robust.
In conclusion, we demonstrated the potential protective mechanism of C6orf120 against CCl4-induced hepatic damage. The hepatoprotective effects of C6orf120 are associated with the downregulation of NLRP3 inflammasome and JNK signaling pathways. This study revealed the regulatory role of C6orf120 in the pathogenesis of ALI, highlighting a potential therapeutic target.