Introduction
Ischemic heart disease is characterized by high morbidity and mortality and is a major cause of death worldwide [1]. Although rapid reperfusion is considered the most effective therapy for myocardial infarction [2], it can induce injury during the early stages, termed myocardial ischemia/reperfusion (I/R) injury. It was reported that I/R injury disturbs ionic homeostasis, increases the production of reactive oxygen species (ROS) and inflammatory responses, induces mitochondrial disorders, and causes calcium overload [3].
Autophagy is a crucial house-keeping process that controls the degradation of long-lived proteins and damaged organelles, playing a vital role in I/R in the myocardium [4]. Autophagy is induced in response to stress conditions in the heart, including cardiac muscle hypertrophy and heart failure, and is regarded as a prominent feature of cardiovascular dysfunction [5–11]. It has been shown that autophagy exerts a dual function during I/R injury in the myocardium. On one hand, autophagy compensates for mitochondrial injury, providing support to proteostasis and the survival of damaged cardiomyocytes following I/R injury [12–14]. However, sustained autophagy during reperfusion can induce cell death through the digestion of essential proteins and organelles. Matsui et al. indicated that autophagy plays a protective role during ischemia, but can detrimentally affect the heart during reperfusion [15]. Regarding the early reperfusion of ischemic myocardium, a modest increase in autophagy protects against necrocytosis due to the degradation of damaged organelles and the provision of free fatty acids (FFA) and amino acids, but sustained up-regulation of autophagy drives cardiac cell apoptosis and/or injury, leading to cell death. In addition, autophagy is an important regulator of glucose homeostasis. He et al. revealed that autophagy controlled by exercise-induced Bcl-2 is essential for muscle glucose homeostasis, and that Bcl-2-knockout mice fail to increase GLUT4 localization at the plasma membrane, reducing glucose uptake [16]. Roy et al. revealed that TBC1D5 shuttling is dependent on autophagy as it controls the plasma membrane translocation of GLUT1 [17].
It has been reported that dichloroacetate (DCA) activates pyruvate dehydrogenase and stimulates glucose aerobic metabolism in perfused hearts [18, 19]. DCA also reduces oxygen consumption and lactate production [20]. These data demonstrate that DCA ameliorates cardiac dysfunction by depleting lactic acidosis [21]. Stacpoole et al. [22] reported that DCA elevates systolic pressure and cardiac output for those suffering from hypotension and lactic acidosis. Moreover, Burns et al. [23] suggested that DCA stimulates pyruvate dehydrogenase and restores myocardial ATP levels to augment myocardial protection in rat hearts. However, the role of DCA in I/R through its ability to regulate autophagy has not been reported.
In this study, we hypothesized that DCA may alleviate I/R injury through the inhibition of autophagy. We further investigated the potential mechanism(s) involved in the regulation of autophagy by DCA.
Material and methods
Myocardial ischemia-reperfusion model in vivo
Healthy Sprague-Dawley (SD) rats weighing 250–300 g were purchased from the Institute of Laboratory Animal Sciences, Peking Union Medical College (Beijing, China). Rats were raised individually in plastic cages under conditions of a 12 h light/dark cycle (temperature at 23 ±1°C; 12 h light/dark cycle, light on at 7 a.m.). Thirty SD rats were randomly divided into three groups: the control group (normal saline), the I/R group (normal saline and I/R) and the I/R + DCA (3 mM) group. Each group consisted of 12 rats. The study group was anesthetized with 3% sodium pentobarbital (i.p.) (60 mg/kg) and treated with heparin (i.p.) (150 U/kg). The rats in the control group underwent a sham operation. Myocardial I/R was produced in rats of the I/R group according to previous studies [24]. Briefly, the BIOPAC multi-guide biometric device was used to test the ECG with needle electrodes installed under the skin. A catheter was inserted into the carotid artery to take blood samples upon completion of the experiment. Following intubation, rats were ventilated mechanically using a rodent ventilator (SAR-830, USA). The chest was opened, the pericardium was incised, and the heart was visible. Subsequently, the left anterior descending coronary artery was ligated to perform coronary artery occlusion, which was confirmed by ST segment elevation, an increase in R-wave amplitude and epicardial cyanosis. At 30 min post-occlusion, a snare was used to release the coronary artery occlusion, and the heart was reperfused for 120 min. Additionally, reperfusion was confirmed by the recovery from cyanosis and the inversion of ECG changes [25]. After reperfusion, hearts were separated and stained with 1% (w/v) triphenyl tetrazolium chloride (TTC) to assess infarct size. All procedures were strictly conducted in accordance with the national (D.L.n.26, March 4, 2014) and international regulations and policies (directive 2010/63/EU). This protocol was permitted by the Animal Experimental Ethics Committee, Beijing Chao-Yang Hospital, Capital University of Medical Science.
Evaluation of cardiac function
Arrhythmia was defined and determined based on the criteria of the Lambeth Conventions [26]. ECG recordings were analyzed during the first 30 min of reperfusion for ventricular arrhythmias. Arrhythmias were categorized into 6 groups and their severity scored [27]: 0 = no arrhythmia; 1 = less than 10 s of ventricular tachycardia or other arrhythmias, with no ventricular fibrillation (VF); 2 = 11 s to 30 s of ventricular tachycardia or other arrhythmias, with no VF; 3 = 31 s to 90 s of ventricular tachycardia or other arrhythmias, with no VF; 4 = 91 s to 180 s of ventricular tachycardia or other arrhythmias, and/or less than 10 s of reversible VF; 5 = greater than 180 s of ventricular tachycardia or other arrhythmias, and/or greater than 10 s of reversible VF; 6 = irreversible VF. In addition, the heart rate (HR), the left ventricular developed pressure (LVDP), and the maximal rate of pressure rise (+dp/dtmax) were recorded, and the coronary flow (CF) was measured to evaluate cardiac function.
Serum determination
After 120 min of reperfusion, 3 ml of blood was collected through the common carotid artery to detect the levels of serum creatine kinase (CK) (Abcam), lactic dehydrogenase (LDH) (Abcam) and cardiac troponin I (cTnI) (Abcam) by enzyme-linked immunosorbent assay (ELISA). Assays were performed according to the manufacturer’s instructions.
Cardiac myocyte culture
Cardiac myocytes in neonatal rats were isolated and maintained as previously described [28]. Briefly, isolated cells were cultured in serum-free, glucose-free Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific, Shanghai, China) in the anoxia chamber (95% N2, 5% CO2) at 37°C for 24 h. Reperfusion was then initiated through the replacement of normal culture media containing 10% fetal bovine serum (FBS) (Gibco) in 5% CO2 and 95% O2 for 2 h. At the beginning of the reperfusion, different concentrations of DCA were used to treat cells and were maintained for 12 h. Cells were then harvested for specific assays.
Myocardial cell viability and apoptosis
To gain insight into the role of DCA in cardiac myocytes, cell viability and apoptosis assays were performed. Treated myocardial cells were seeded into 96-well plates at a density of 10,000 cells per hole and viability was assessed via MTT assay. For the assessment of apoptosis, TUNEL staining was performed according to the manufacturer’s instructions. Apoptotic cells were counted in five random visual fields. The percentage of cells undergoing apoptosis was calculated using the following equation: Apoptosis % = number of apoptotic cells/total number of apoptotic cells × 100%.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA isolation from myocardial cells was performed to analyze the expression of Bcl-2 and Bax at the mRNA level using TaqMan assays (Life Technologies, CA, US). Tests were performed as previously described [29].
Immunofluorescence
For immunofluorescence, cardiac myocytes were first fixed with 4% paraformaldehyde (PFA) for 10 min. Cardiac myocytes were then permeabilized with 0.1% Triton X-100 for 10 min, blocked for 1 h with 1% bovine serum albumin (BSA), and incubated with anti-LC3 antibody (Abcam, dilution 1 : 100) and anti-Beclin-1 antibody (Abcam, dilution 1 : 100) for 12 h at 4°C. After incubation of primary antibodies, cardiac myocytes were incubated with anti-mouse IgG labeled with Alexa Fluor-488 for 1 h at 37°C and then stained with DAPI for 5 min. The immunofluorescence results were assessed under Laser confocal microscopy (Carl ZEISS, Oberkochen, Germany).
Intracellular ROS detection
The myocardium of the left ventricle was sectioned and fixed in 10% formalin, and frozen in liquid nitrogen prior to use. Assays were performed as previously described [30]. ROS positive cells were detected by DCFH-DA (Beyotime, China). Using fluorescence microscopy, cell images were obtained and ROS fluorescence intensity was quantified using Image J software (NIH, Bethesda, Maryland, USA).
siRNA treatment
Hairpin siRNA was used to silence Bcl-2 expression in myocardial cells. siRNA was synthesized by Thermo Fisher. Bcl-2 siRNA: 5′-GGGAGAACAGGGTACGATA-3′, scrambled control siRNA: 5′-GGGAAACGGTGCAGAGATA-3′. Following the transient transfection of myocardial cells with siRNA, cells were incubated under normal conditions. Samples were then prepared for further assays.
Western blot
Total proteins from cultured cells and the left ventricle were extracted according to our protocols. Western blot assays were conducted as previously described. Obtained proteins were incubated with anti-Beclin-1 (1 : 1000, Cell Signaling Technology), anti-Bcl-2 (1 : 1000, Cell Signaling Technology), anti-LC3 (1 : 1000, Cell Signaling Technology), anti-Glut1 (1 : 1000, Abcam) and anti-Glut4 primary antibodies (1 : 100, Abcam).
Statistical analysis
All data analysis was performed using SAS 9.3 software (SAS Institute, USA) and a p-value less than 0.05 was considered statistically significant. Normally distributed data with homogeneous variance were expressed as the mean ± standard deviation (SD). A one-way analysis of variance was performed for multiple group comparisons, followed by post-hoc analysis for group-wise comparisons.
Results
Effects of DCA on the myocardial I/R model in vivo
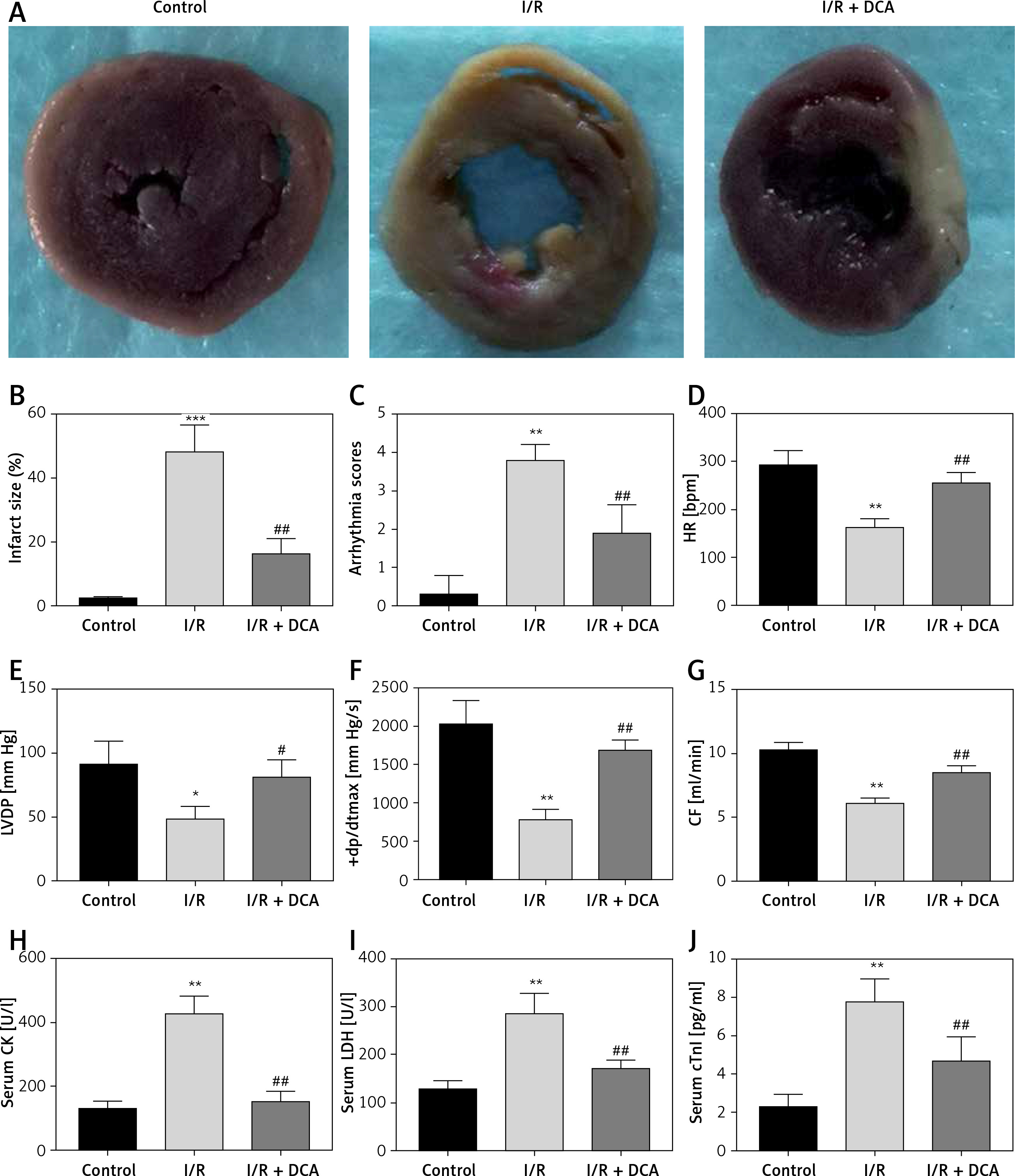
To assess whether DCA can alleviate myocardial I/R injury, we compared the cardiac infarct size, cardiac arrhythmia scores, HR, LVDP, +dp/dtmax, CF and serum myocardial injury markers during reperfusion in the three experimental groups. The infarct size in the I/R group was significantly higher than the control group, while DCA significantly reduced the infarct size compared to the I/R group (Figures 1 A, B). Notably, arrhythmia scores in the I/R groups were dramatically higher in comparison to the control group, but treatment with DCA strongly reduced the arrhythmia score, suggesting that it reverses the effects of IR (Figure 1 C). The HR, LVDP, +dp/dtmax, and CF were significantly lower in the I/R group compared to the control group, while they were significantly higher after treatment with DCA compared to the I/R group (Figures 1 D–G). In addition, we also observed an apparent increase in the serum levels of CK, LDH and cTnI in the I/R group, which was reversed by DCA treatment (Figures 1 H–J). Collectively, these results confirmed that DCA exerts a protective role in myocardial I/R models in vivo.
Figure 1
Evaluation of arrhythmia and myocardial damage indicators in vivo. A – TTC staining results of hearts in control, ischemia/reperfusion (I/R) and I/R+ dichloroacetate (DCA) groups (n = 4); B – infarct size of hearts in the three groups (n = 4); C – arrhythmia score changes in the three groups (n = 4); D – heart rate (HF) changes in the three groups (n = 4); E – left ventricular developed pressure (LVDP) changes in the three groups (n = 4); F – the maximal rate of pressure rise (+dp/dtmax) changes in the three groups (n = 4); G – coronary flow (CF) changes in the three groups (n = 4); H – I/R groups display higher creatine kinase (CK) levels compared to the control group (n = 4); I – serum lactic dehydrogenase (LDH) (n = 4); J – serum cardiac troponin I (cTnl) (n = 4). I/R represents ischemia/reperfusion group. Rats in the I/R + DCA group had ischemia/reperfusion and were treated with DCA
**P < 0.01 compared to the control group. ## P < 0.01 compared to the I/R group.
In vitro effects of DCA on myocardial cell viability and apoptosis
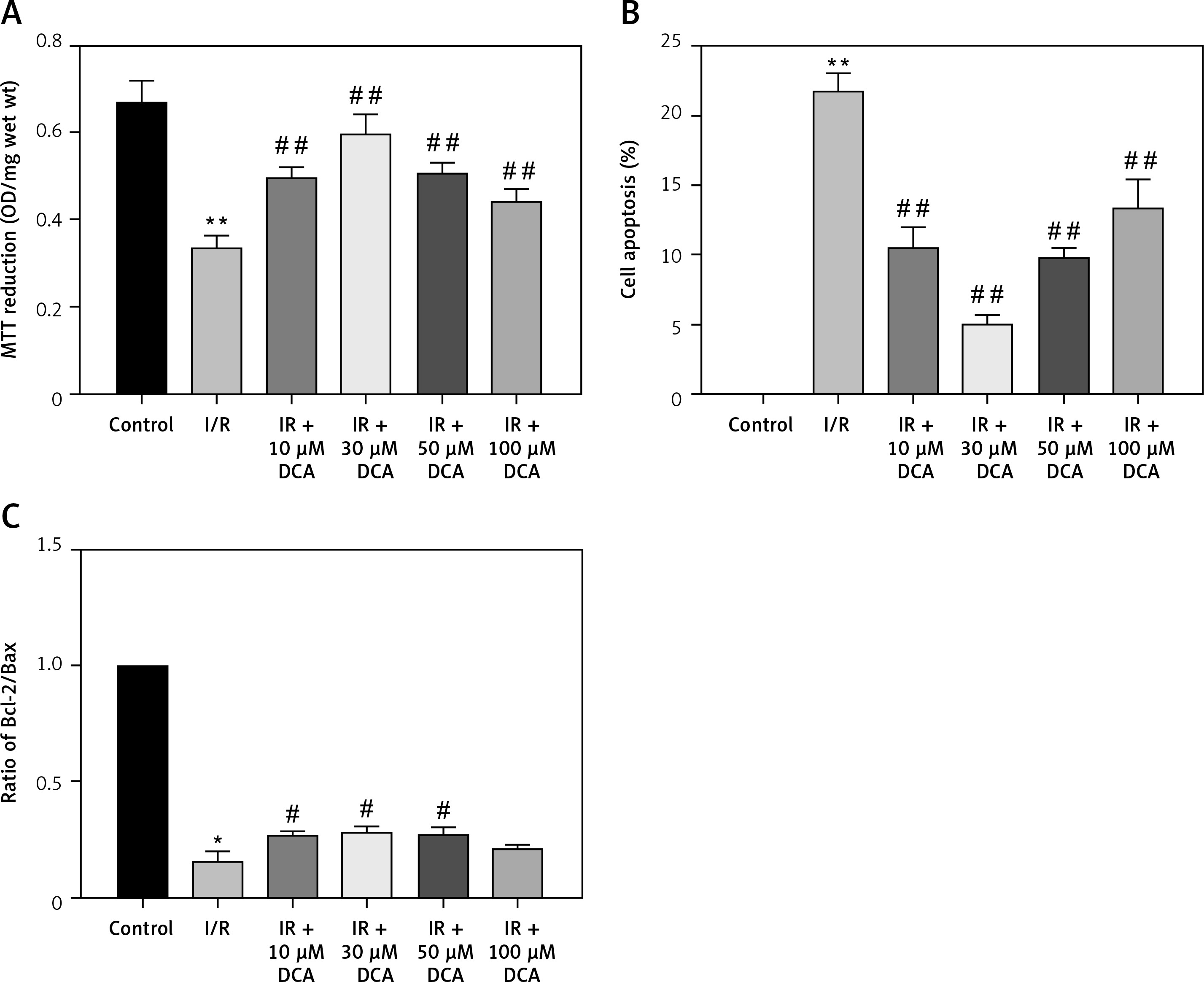
We next determined the viability of myocardial cells in vitro through MTT assays. Treated cells were divided into control, I/R, and I/R + DCA groups. The results showed that cell viability in the I/R group was lower than the control group (p < 0.01) (Figure 2 A). Notably this effect was reversed by DCA treatment. When the concentration of DCA was 30 μM, cell viability improved (Figure 2 A). When myocardial cell apoptosis was determined by TUNEL assay, the percentage of apoptotic cells in the I/R group was strongly elevated compared to the control group. It was noteworthy that DCA treatment reversed these effects and 30 μM DCA significantly alleviated cell apoptosis (Figure 2 B). In addition, when the expression of Bcl-2 and Bax was assessed at the mRNA level, qRT-PCR assays indicated that the Bcl-2/Bax ratio in the I/R group was strongly down-regulated, whilst DCA recovered the Bcl-2/Bax ratio (Figure 2 C). Taken together, these observations demonstrate that DCA promotes cell viability and inhibits apoptosis following I/R.
Figure 2
Effects of dichloroacetate (DCA) on myocardial cell viability and apoptosis in vitro. A – Cell viability was determined by MTT assays in the six groups (n = 4); B – effects of DCA on cell apoptosis induced by ischemia/reperfusion (I/R) assessed through TUNEL staining (n = 4); C – expression of Bcl/Bax in each group (n = 4).
**P < 0.01 compared to control group; ##p < 0.01 compared to the I/R group.
DCA attenuates I/R injury by suppressing reperfusion-induced autophagy
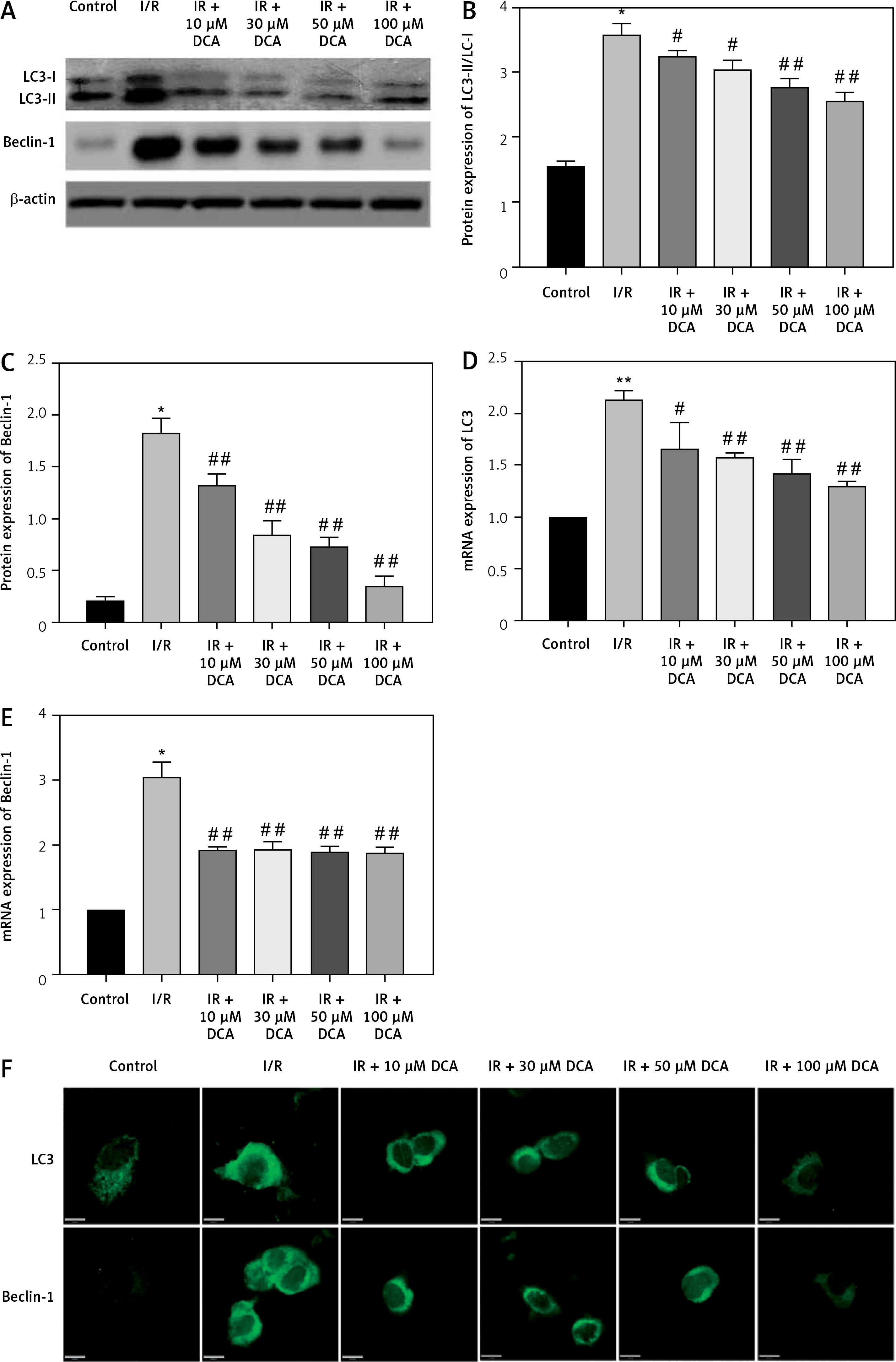
To explore the role of DCA in the regulation of autophagy, we analyzed its effects on Beclin-1 and LC3-II expression, considered reliable autophagy biomarkers. Western blot analysis indicated that Beclin-1 level (p < 0.05, Figures 3 A, B) and the ratio of LC3-II/LC3-I (p < 0.05, Figures 3 A, C) were significantly higher in the I/R group in comparison to the control group. Conversely, this effect was significantly inhibited in a concentration-dependent manner by DCA treatment (Figures 3 A–C). These findings were confirmed at the mRNA level through qRT-PCR (Figures 3 D, E). In addition, the immunofluorescence results also showed that the expression of LC3 and Beclin-1 increased in the I/R group and decreased in the DCA treated group (Figure 3 F). Collectively, these results revealed that DCA inhibits reperfusion-induced autophagy through the down-regulation of potential indicators.
Figure 3
Dichloroacetate (DCA) attenuates ischemia/reperfusion (I/R) injury by suppressing reperfusion-induced autophagy. A – Western blot analysis of LC3-II/LC3-I and Beclin-1 following DCA treatment (n = 4); B – relative expression of LC3-II/LC3-I (n = 4); C – Beclin-1 expression was determined by western blot (n = 4); D – LC3, E – Beclin-1 mRNA levels in the six groups (n = 4); F – immunofluorescence results of LC3 and Beclin-1 in the six groups (n = 4)
*P < 0.05 compared to control group. #P < 0.05 compared to I/R group. ##P < 0.01 compared to I/R group.
Effects of DCA on autophagy through ROS and Bcl-2
It has been reported that ROS accumulation drives the oxidative destruction of DNA, proteins and lipids, triggering cell damage and apoptosis [24]. ROS accumulation is also essential for autophagy induction under conditions of starvation or stress [25]. To assess the effects of ROS in the DCA-mediated inhibition of autophagy, we assessed the levels of ROS in myocardial cells using DCFH-DA. The results showed that ROS production increased after I/R, which was significantly reduced by DCA (Figure 4 A). These effects were lost following treatment with N-acetyl-l-cysteine (NAC), a classical ROS inhibitor.
Figure 4
Effects of dichloroacetate (DCA) on autophagy through reactive oxygen species (ROS) and Bcl-2. A – ROS generation was detected by DCFH-DA in myocardial cells (n = 4); B – expression levels of Bcl-2, LC3-II/LC3-I and Beclin-1 after Bcl-2 siRNA silencing in ischemia/reperfusion (I/R) + DCA groups (n = 4); C – band intensities from western blots were calculated and normalized to GAPDH levels (n = 4). Protein expression is expressed as the fold change relative to experimental controls
*P < 0.1, **p < 0.01.
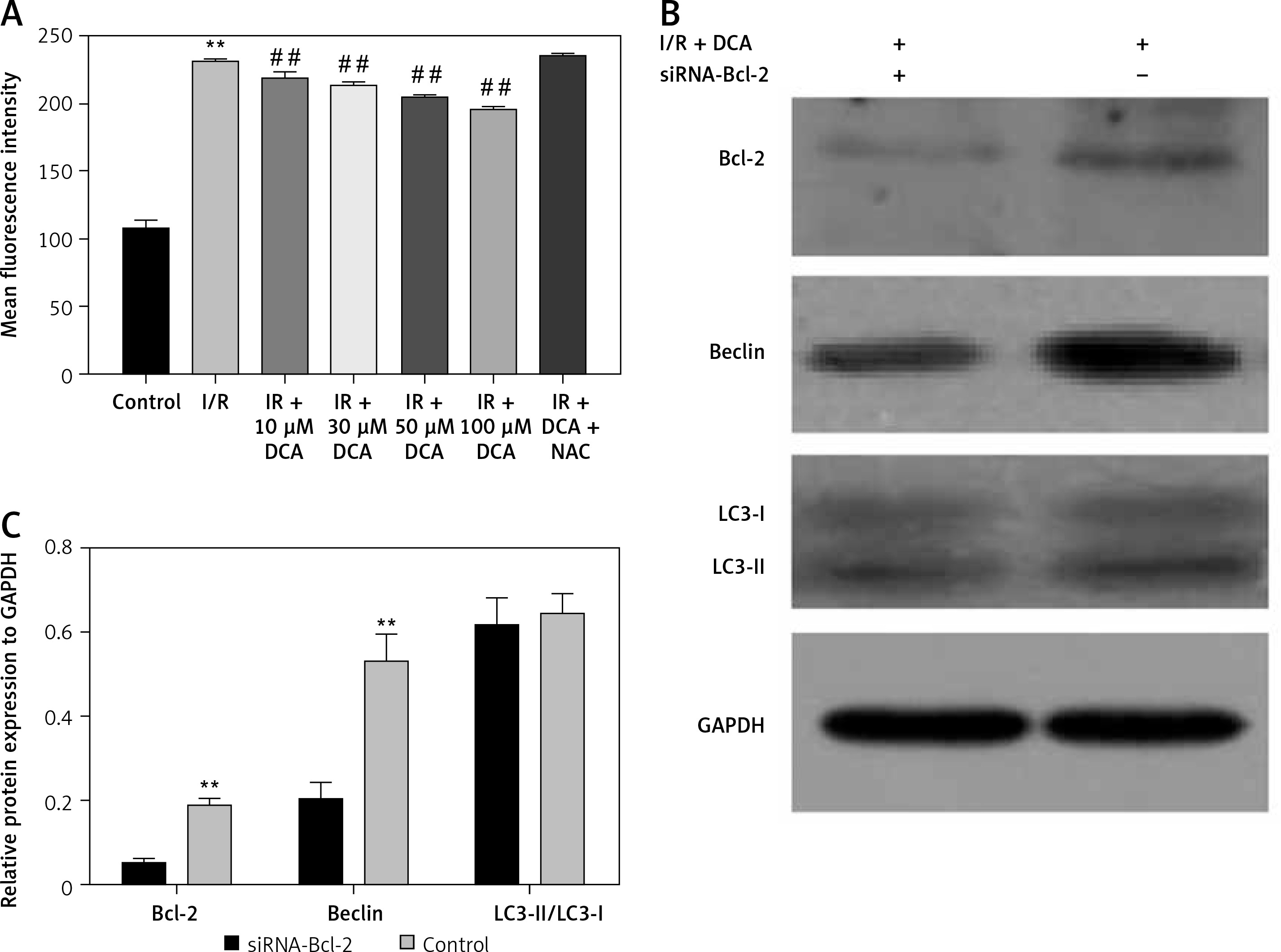
Hairpin siRNA was used to silence Bcl-2 and western blot analysis revealed that Beclin-1 expression decreased following DCA treatment. A significant difference in the LC3-II/LC3-I levels following Bcl-2 knock-down was not observed (Figures 4 B, C). These observations suggest that ROS and Bcl-2 contribute to DCA-mediated autophagy inhibition.
Effects of DCA on myocardial glucose homeostasis in vivo
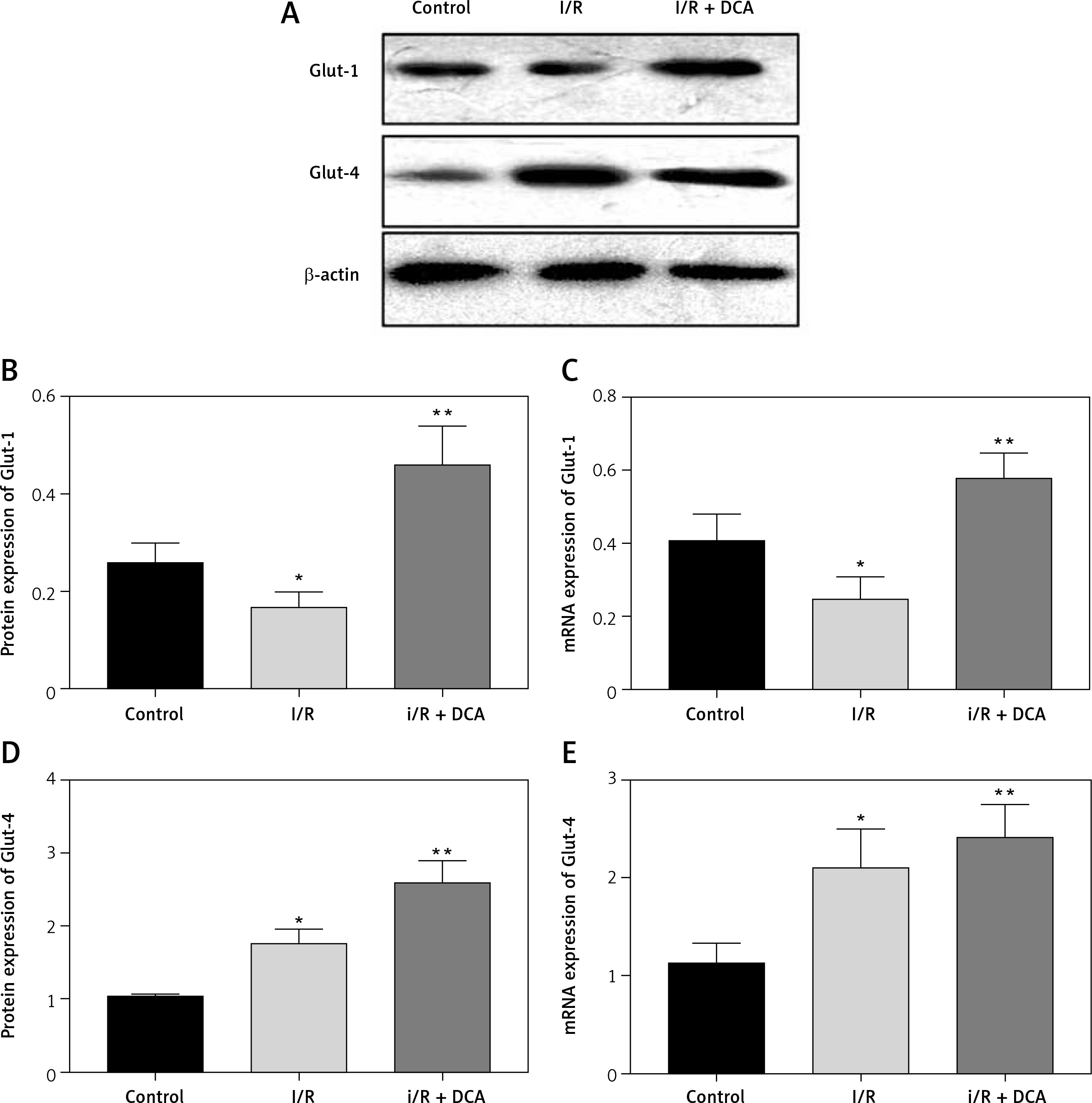
We finally assessed the effects of DCA on myocardial glucose homeostasis after reperfusion in vivo. Data from both western blot and qRT-PCR assays revealed upregulation of Glut-1 and Glut-4 at both the protein and mRNA level in left ventricular tissue following DCA treatment Figures 5 A–E. This suggested that DCA promotes glucose uptake.
Figure 5
Effects of dichloroacetate (DCA) on myocardial glucose homeostasis in vivo. Glut-1 (A, B) and Glut-4 (A, C) expression levels were detected by western blot analysis in the three groups. Glut-1 (D) and Glut-4 (E) mRNA levels were also determined by RT-PCR (n = 4)
*P < 0.05 compared to the control group. **P < 0.01 compared to the control group.
Discussion
Ischemic heart disease involving myocardial infarction leads to serious health issues. Rapid reperfusion of the ischemic myocardium remains the fundamental therapy for myocardial infarction [2]. Importantly, an additional injury, termed myocardial I/R, occurs during the initial period. DCA can activate pyruvate dehydrogenase and improves cardiac function [31]. In this study, we explored the functional implications of DCA after I/R injury in vitro and in vivo and identified a protective role of DCA against reperfusion injury through the modulation of autophagy and glucose homeostasis.
Autophagy is an intracellular bulk degradation process that has the capacity to degrade and recycle cytosolic and long-lived proteins and organelles [4, 32, 33]. Autophagy occurs at low levels and is enhanced by stressors such as nutrient depletion [34]. Recent studies have shown that autophagy has both beneficial and adaptive roles depending on the context and its magnitude of induction [35]. Autophagy is also implicated in I/R injury. At low levels, autophagy can prevent cell injury following I/R. In high concentrations or through sustained up-regulation, autophagy can promote cardiac cell apoptosis or cell injury through Beclin-1 [36]. This study indicated that DCA improves cardiac function, increases cell viability and inhibits apoptosis. Furthermore, the first evidence that DCA can inhibit Beclin-1 expression and influence the LC3-II/LC3-I ratio is provided, suggesting that DCA reduces autophagy induction during the reperfusion period.
Growing evidence demonstrates that ROS-activated autophagy occurs during myocardial reperfusion [37, 38]. ROS functions as an essential signaling molecule that modulates a series of biological processes, including gene expression, cell metabolism, growth and survival. However, excessive ROS is detrimental to cellular homeostasis, leading to cell damage and apoptosis [24, 39]. It has been reported that autophagy is upregulated through ROS accumulation during myocardial reperfusion. In this study, we found that DCA treatment strongly inhibited ROS production during myocardial reperfusion, but NAC (an ROS inhibitor) had the capacity to abolish this effect. Combined with the aforementioned data, these results indicate that ROS contributes to DCA-mediated autophagy inhibition, promoting myocardial cell protection during the reperfusion period.
It has been demonstrated that Bcl-2 is involved in apoptotic signaling. Interestingly, Bcl-2 family members regulate a variety of biological effects, including morphogenesis, glucose homeostasis, and autophagy [16, 40, 41]. Moreover, previous studies have shown that Bcl-2 decreases the levels of starvation-induced autophagy in vitro and in vivo, indicating its key role in autophagy regulation [42, 43]. In this in-vitro study, DCA significantly alleviated apoptosis by up-regulation of the Bcl-2/Bax ratio. Following Bcl-2 silencing, Beclin-1 was inhibited by DCA treatment, suggesting that it contributes to the ability of DCA to regulate autophagy. We also found that Bcl-2 silencing does not influence the LC3-II/LC3-I ratio, a finding that warrants further investigation.
It was recently reported that myocardial efficiency is reduced when the levels of FFA are elevated [44, 45]. We therefore predict that myocardial glucose abundance decreases oxygen consumption to improve myocardial efficiency. In addition, autophagy regulates glucose homeostasis [16]. We found that DCA dramatically enhanced Glut-1 and Glut-4 expression, raising the possibility that DCA represses autophagy to improve myocardial energy metabolism.
In conclusion, DCA regulates autophagy and glucose homeostasis to improve I/R injury. Importantly, we revealed the utility of DCA as a therapeutic strategy for I/R injury.