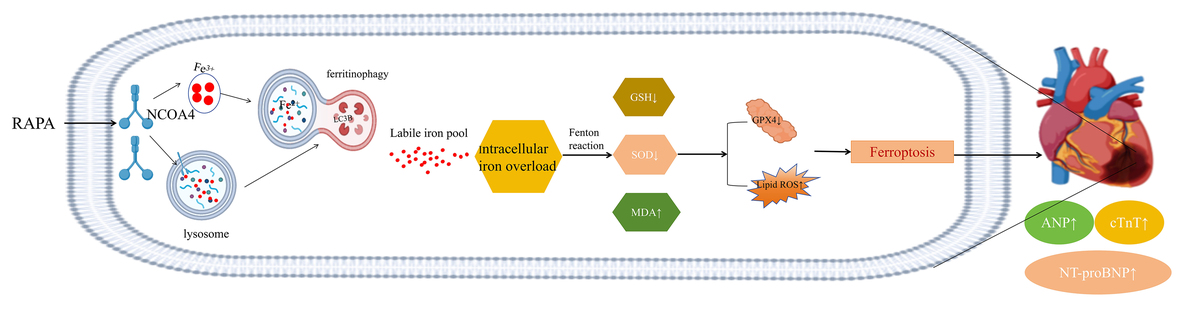
Introduction
Acute myocardial infarction (AMI) is associated with significant mortality and morbidity. It may cause early cardiac remodeling that leads to cardiac decompensation and heart failure (HF) as a result of inadequate blood and oxygen supply to the heart muscle [1–3]. According to the latest epidemiological data, there are about 12.1 million heart failure patients in China among people aged 25 years and above, and about 3 million new heart failure patients each year [4]. Neuroendocrine disorders, immune response activation, oxidative stress, and lifestyle factors such as obesity and anxiety contribute to the complex, multifactorial pathogenesis of HF [5]. However, the precise pathophysiological mechanisms underlying post-MI HF have not yet been fully elucidated.
Autophagy is an evolutionarily conserved mechanism that plays a vital role in cellular metabolism, renewal, and maintenance of cell survival, differentiation, development, and homeostasis through lysosome-mediated cellular degradation [6]. In 2014, Mancias et al. introduced the term “ferritinophagy” to describe this phenomenon of selective autophagy against ferritin. In this process, NCOA4 acts as a cargo receptor that mediates ferritin degradation and releases free iron [7]. NCOA4 is present in autophagic lysosomes and facilitates the recognition and targeting of ferritin by autophagic vesicles through its C-terminal helical structural domains [8]. By binding to microtubule-associated protein 1A/1B light chain 3-PE(LC3-PE) and FTH1 on the autophagosome membrane, NCOA4 isolates iron-containing ferritin complexes within the autophagosome. Upon maturation and fusion of the autophagosome with the lysosome, NCOA4-mediated ferritin is degraded, leading to the release of bioavailable iron [9].
While free iron is physiologically involved in various biological processes, excessive levels can promote ferroptosis [10]. Ferroptosis is an iron-associated form of cell death whose main features include reduced intracellular expression of glutathione (GSH), decreased activity of GPX4, and subsequent accumulation of reactive oxygen species (ROS) and lipid peroxidation [11]. Abnormal iron metabolism, including ferroptosis, has been closely associated with the development of cardiovascular diseases such as atherosclerosis [12], HF [13, 14], myocardial ischemia/reperfusion injury [15], and cardiomyopathy [16, 17].
As ferritinophagy maintains iron homeostasis, its dysregulation may lead to iron overload and ferroptosis [18]. Ferritin is a cytosolic iron storage protein complex with substantial antioxidant activity as it chelates redox-active iron. It is composed of 24 heavy (ferritin heavy chain 1 – FTH1) and light (ferritin light chain – FTL) subunits [14]. NCOA4-mediated ferritinophagy might be implicated in cardiovascular disease (CVD) pathogenesis [14]. The inhibition of 3-methyladenine (3-MA) and the autophagy-associated gene ATG5 (autophagy-related 5) blocked ferroptosis and prevented lipopolysaccharide (LPS)-induced cardiac injury [19]. Hence, there may be a strong association between autophagy and ferroptosis. Autophagy is an upstream mechanism that induces ferroptosis by regulating cellular iron homeostasis and ROS production, while ferritinophagy could serve as a bridge between autophagy and ferroptosis [20]. Our previous study demonstrated the importance of autophagy in HF pathogenesis. In cardiomyocytes, normal autophagy favors survival whereas excessive autophagy promotes necrosis [21–23]. However, the mechanisms by which autophagy influences ferritinophagy, ferroptosis, and post-MI HF development remain unclear. Therefore, the objectives of the present study were to (a) determine whether ferritinophagy links autophagy and ferroptosis, and (b) elucidate the molecular mechanisms by which autophagy induces HF by promoting NCOA4-mediated ferritinophagy, releasing free iron, inhibiting GPX4, and accelerating ferroptosis.
Material and methods
Experimental animals
Forty-eight-week-old male Sprague-Dawley (SD) rats weighing between 200 and 250 g were obtained from Zhengzhou Huaxing Experimental Animal Breeding Co. Ltd. (license number: SCXK (YU) 2019-0002). The experimental protocol adhered to the guidelines outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals [24]. The SD rats were randomly divided into four groups: CON (normal group), MOD (post-MI HF), RAPA (rapamycin (1 mg/kg, q.d. 4w), and 3-MA (3-methyladenine (15 mg/kg, q.d. 4w)) groups. Except for the CON group, all groups underwent left coronary artery ligation.
Animal model
Prior to surgery, the rats were anesthetized and intubated, and a small animal anesthesia ventilator was utilized. Anesthesia was induced using isoflurane (20201202, Shandong Ante Herding Technology Co., Ltd., China) at a concentration of 3–4% and maintained at a concentration of 1.5–3%. A 6-0 silk suture was passed through the left coronary trunk and ligated along with a small bundle of myocardium, resulting in visible lightening and paleness below the suture [25]. The thoracic cavity was closed using conventional sutures, and intraperitoneal injection of furosemide (20230106, Hefei Zhonglong Shenli Animal Pharmaceutical Co., Ltd., China) at a dose of 0.2 ml per animal was administered after thoracic closure. After 4 weeks of regular feeding, echocardiography was performed to identify rats with left ventricular ejection fractions (LVEF) values below 60% and fractional shortening (FS) values below 30%, establishing them as the post-myocardial infarction HF model and assigning them to the respective groups [22]. At the end of the experiment, euthanasia was performed by intraperitoneal injection of 3% sodium pentobarbital (30 mg/kg).
Echocardiographic assessment
Anesthesia was induced and maintained using isoflurane (20201202, Shandong Ante Herding Technology Co., Ltd., China) at concentrations of 3–4% and 1.5–3%, respectively. Two-dimensional B-mode and M-mode echocardiography (Siemens Acuson Oxana 3; Siemens AG) were utilized to assess left ventricular cardiac function. Chest echocardiography was performed during the fifth week to identify the model.
Pathological staining
Heart samples were fixed in 4% paraformaldehyde at 4°C for 48 h. Subsequently, they were dehydrated through a series of ethanol solutions with increasing concentrations, clarified in xylene, and embedded in paraffin. Paraffin-embedded tissue sections were prepared and subjected to hematoxylin and eosin (H&E) staining to assess histopathological features. The paraffin sections were dewaxed using xylene I and II, followed by a graded ethanol and alcohol wash, and finally rinsed with distilled water. The sections were then stained with hematoxylin for 40 s and eosin for 3 s, and washed with tap water and distilled water sequentially. Myocardial morphological changes were observed, and image information was captured using an optical microscope.
Immunofluorescence staining
Heart tissues were fixed in paraffin and incubated overnight at 37°C. The paraffin-embedded heart sections were then deparaffinized and treated with 3% H2O2 for 20 min. After rinsing with PBS, the sections were incubated with primary rabbit anti-NCOA4 antibody (1 : 100, DF4255, Affinity Biosciences, China) and primary rabbit anti-GPX4 antibody (1 : 100, DF4255, Affinity Biosciences, China) separately at 37°C for 60 min. Immunofluorescence co-localization was achieved by incubating the sections with the primary antibody NCOA4 for 60 min, followed by the addition of the corresponding secondary antibody (Cy3). Subsequently, the sections were incubated with the second primary antibody LC3 (1 : 300, AF4650, Affinity Biosciences, China) and the corresponding secondary antibody (FITC, 1 : 200, S0008, Affinity Biosciences, China). The slides were then sealed with an anti-fluorescence quencher containing DAPI and examined under a fluorescent inverted microscope (Olympus, Japan) at 200× magnification.
Transmission electron microscopy
Multiple myocardial tissue pieces, each measuring 1 mm3, were fixed in a 2.5% glutaraldehyde solution. They were subsequently dehydrated through an ethanol gradient, infiltrated with epoxy resin for 2–3 h, and embedded in pure epoxy resin. After trimming, the samples were baked at 40°C for 12 h and then at 60°C for 48 h. Ultra-thin sections with a thickness of 70 nm were cut, transferred onto copper grids, and stained with lead and uranium for the purpose of observing mitochondrial morphology and autophagic bodies using transmission electron microscopy.
Cell culture
H9C2 cells (1 × 105 cells, iCell-r012, iCell, China) were cultured in DMEM (G4512, Solebro, China) supplemented with 10% fetal bovine serum and 100 U/ml streptomycin/penicillin. The cells were maintained in a CO2 incubator at 37°C. The cells were seeded in 24-well plates. Short interfering RNA (siRNA) and plasmids were synthesized by Tongyong Biotechnology Co. Transfection of si-NCOA4 (NCOA4 silencing), si-GPX4 (GPX4 silencing), and si-NC (negative control) was performed, and transfection efficiency was assessed using RT-qPCR (Table I). After 6 h of transfection, except for the control group, H9C2 cells in the logarithmic growth phase were subjected to 4 h of hypoxia in serum-free medium in a hypoxia chamber (37°C, 5% CO2, 95% N2). Subsequently, the H9C2 cells were placed in DMEM medium containing 10% FBS and reoxygenated for 4 h to establish the H/R injury model in H9C2 cells [26]. The experimental groups included: CON group (control group), MOD (H/R) group, deferoxamine (DFO) (1 µM, treated for 48 h; D9533, Sigma, USA), ferrostatin-1 (Fer-1) (1 µM, treated for 24 h; HY-100579, MCE, China), necrostatin-1 (Nec) (30 µM, treated for 24 h; HY-15760, MCE, China), and carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (zVAD) (10 µM, treated for 24 h; HY-16658B, MCE, China).
Table I
Sequences of primers used for reverse transcription-quantitative PCR
Cell viability assay
Cell viability was assessed using the cell counting kit 8 (CCK8) assay kit (G4103, Servicebio, China) following the manufacturer’s instructions. H9C2 cells were treated with the CCK-8 solution, which was prepared by diluting 10 ml of CCK-8 solution to the working concentration, and incubated with the cells. After 1 h, the absorbance of each well was measured at OD450 nm using an ELISA reader.
Catalytic Fe (II) imaging
H9C2 cells (1 × 105 cells) were seeded in 24-well culture plates and incubated overnight. The cells were then co-incubated with 5 µM FeRhoNox-1 probe (MX4558, Mao Hong Biotechnology, China) and 50 nM LysoTracker green DND-E26 (MX4318, Mao Hong Biotechnology, China) at 37°C and 5% CO2 for 60 min. Images were captured using a fluorescent inverted microscope (Olympus, Japan) at a magnification of 200×.
Measurement of lipid peroxidation in vitro
H9C2 cells (1 × 105 cells) were seeded in 24-well culture plates and incubated overnight. The cells were then treated with C11 BODIPY 581/591 (2.5 µM) (GC40165, GLPBIO, USA) in growth medium for 30 min. Fluorescence of C11 BODIPY was measured by simultaneously acquiring green (484/510 nm) and red signals (581/610 nm) to assess the rate of lipid peroxidation.
Determination of intracellular labile iron pool (LIP) using FeRhoNox-1 fluorescent probe
H9C2 cells (1 × 105 cells) were seeded in 24-well culture plates and incubated overnight. Subsequently, the cells were incubated with 5 µM Fe-RhoNox-1 probe (MX4558, Mao Hong Biological, China) at 37°C with 5% CO2 for 60 min. After washing with PBS, the cells were observed and photographed under a fluorescent inverted microscope (Olympus, Japan) at a magnification of 200×.
Flow cytometry determination of ROS and lipid reactive oxygen species (lipid ROS)
H9C2 cells (1 × 105 cells) were seeded in 24-well culture plates and incubated overnight. For the measurement of lipid ROS, the C11 BODIPY 581/591 lipid peroxidation fluorescent probe (GC40165, GLPBIO, USA) was added to the cells at a final concentration of 2.5 µM and incubated for 30 min. The cells were then washed three times with serum-free medium. For the measurement of total ROS, the cells were incubated with a working solution of 1 mM dichlorodihydrofluorescein diacetate (DCFH-DA) (MX4802, Mao Hong Biological, China) diluted in DMSO. The cells were suspended in an appropriate amount of the working solution and incubated at 37°C for 30 min in the dark to allow the probe to fully interact with the cells. After incubation, the cells were washed three times with serum-free medium. Flow cytometry analysis was performed using a flow cytometer (BD Accuri C6 Plus, BD Biosciences, USA) and the FlowJo software. The lipid ROS and total ROS levels in individual cells were quantified and analyzed using the Image-Pro Plus 6.0 software (Rockville, USA). The data were normalized to the CON group, which was considered as fold change 1, and the fold changes in ROS levels were calculated.
Relevant indicators for ELISA testing
According to the protocols of the relevant manufacturers, the Creatine kinase-MB (CK-MB) assay kit (RX301189R, Quanzhou Ruixin Biotechnology Co., Ltd., China), N-terminal pro-brain natriuretic peptide (NT-proBNP) assay kit (RX302959R, Quanzhou Ruixin Biotechnology Co., Ltd., China), Iron Assay Kit (I291, DOJinDO, Japan), malondialdehyde (MDA) Assay Kit (A003-1-1, Nanjing Jiancheng Institute of Biological Engineering, China), GSH Assay Kit (A006-2-1, Nanjing Jiancheng Institute of Biological Engineering, China) and superoxide dismutase (SOD) Assay Kit (A003-3-1, Nanjing Jiancheng Institute of Biological Engineering, China) were used. The levels of rat CK-MB, NT-proBNP, ferrous iron (Fe2+), trivalent iron (Fe3+), MDA, GSH, and SOD were measured in H9C2 cells and myocardial tissues.
Western blot assay for protein expression
We extracted lysates from myocardial tissue or cells using RIPA buffer and quantified protein content using the BCA protein assay kit (P0011, Beyotime, China). Twenty µg of protein was subjected to SDS-PAGE electrophoresis (Beyotime, Shanghai, China), transferred to PVDF membrane and blocked in 5% skimmed milk. The membrane was incubated overnight at 4°C with the appropriate primary antibody. The proteins used in the study were as follows: NCOA4 (1 : 500, DF4255, Affinity Biosciences, Jiangsu, China), GPX4 (1 : 500, DF6701, Affinity Biosciences, Jiangsu, China), LC3II/I (1 : 500, AF4650, Affinity Biosciences, Jiangsu, China), cardiac troponin T (cTnT) (1 : 500, DF6261, Affinity Biosciences, Jiangsu, China), atrial natriuretic peptide (ANP) (1 : 500, DF6497, Affinity Biosciences, Jiangsu, China), NT-ProBNP (1 : 500, DF6902, Affinity Biosciences, Jiangsu, China), or β-actin (1 : 2,000, TA-09,Zs Biological Technology, Shanghai, China). Finally, the proteins were incubated with HRP-conjugated secondary antibody (1 : 10000, Proteintech) at room temperature for 2 h. The detection bands were detected using the ECL reagent kit (Beyotime) with an Automatic Exposure Meter (JS-M6P, Shanghai Peiqing Technology Co., Ltd., China) and analyzed using ImageJ software.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
We added 1 ml of TRIzol reagent (Life Technologies, 15596018) to extract total RNA from 1 × 107 cells. Using the PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa, RR047A), and the obtained cDNA was used as a qPCR template. The reaction volume of RT-qPCR was 20 µl, consisting of 0.4 µl of F, 0.4 µl of R, 6.2 µl of RNase Free water, 3 µl of cDNA, and 10 µl of SYBR. QPCR was performed at 95°C for 15 min; 95°C for 10 s, 60°C for 30 s, 40 cycles. The relative quantification (RQ) values of each target gene were calculated using the 2–ΔΔCt method and used for statistical analysis. β-actin was used as an internal reference throughout the entire RT qPCR. The primers used for RT-qPCR are shown in Table II.
Table II
Sequences of primers used for reverse transcription-quantitative PCR
Statistical analysis
Data were statistically analyzed using SPSS 26.0 software (IBM Corp.) and expressed as mean ± standard deviation (SD). Student’s t-test was performed for two-group comparisons. For comparisons of multiple groups, significance was determined using one-way or two-way ANOVA with Tukey’s post-hoc test. If the data were in accordance with the normal distribution and the variance was uniform, one-way analysis of variance was performed. The LSD test was used for the post-hoc comparison of the variance, while the Dunnett T3 method was used for the unequal variance. Data that did not conform to a normal distribution were tested using the nonparametric Kruskal-Wallis H test. P < 0.05 was considered to indicate a statistically significant difference.
Results
Ferritinophagy and ferroptosis were associated with myocardial injury in a rat post-MI HF model
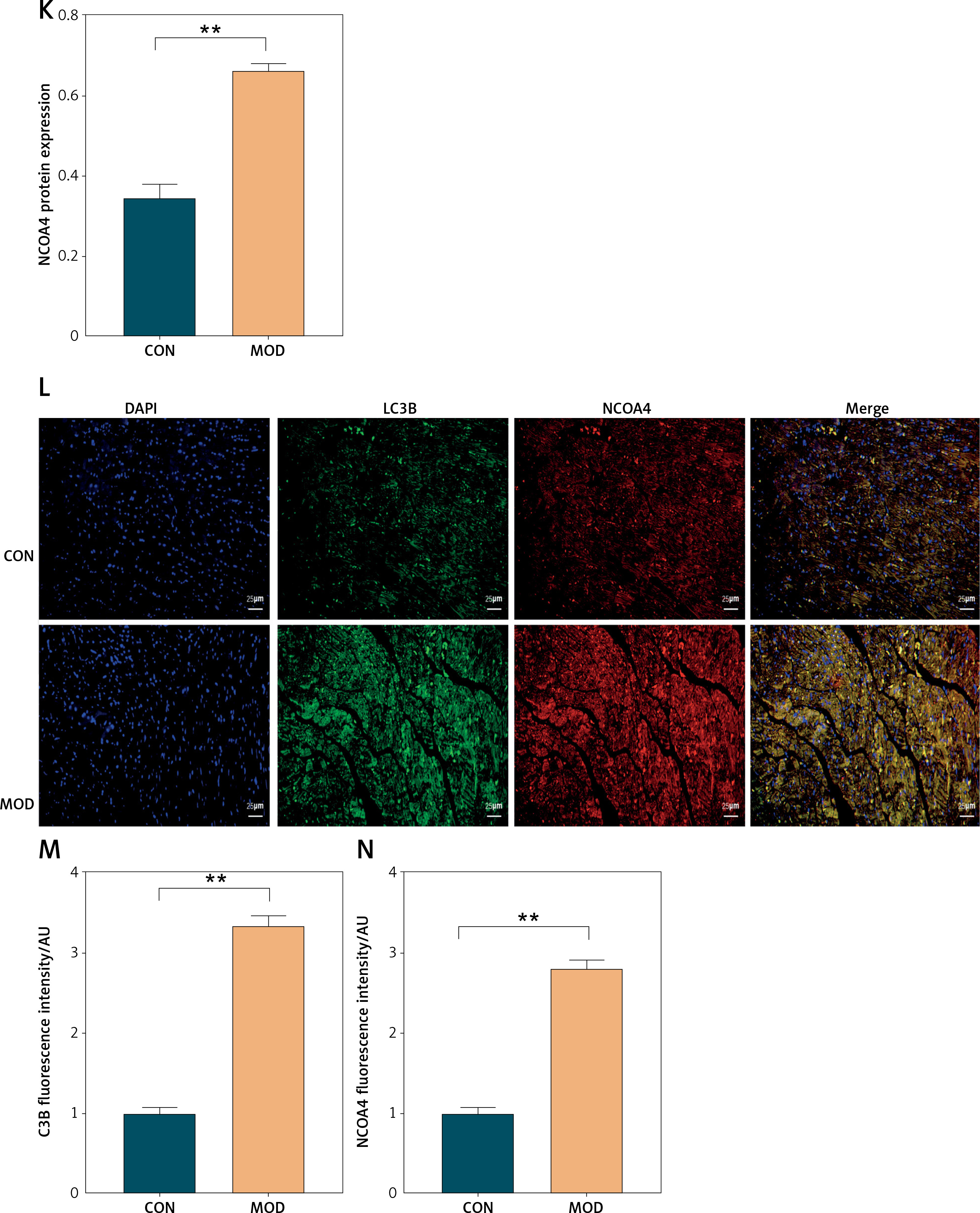
In the present study, we induced post-MI HF in rats by ligating the left anterior descending branch of the coronary artery after 4 weeks. We validated the success of the HF model by evaluating cardiac function with echocardiography. Cardiac function was significantly impaired in the MOD (HF model) group (Figures 1 A–C). Serum ck-MB level exhibited the highest myocardial specificity of all five myocardial enzyme profile indices and was particularly elevated under MI [27]. The ck-MB results were consistent with those of the echocardiography (Figure 1 D). We then measured NCOA4 and GPX4 protein expression to elucidate the mechanisms underlying the observed myocardial injury. NCOA4 mediates ferritinophagy, whereas GPX4 is a key enzyme that protects against ferroptosis by reducing lipid peroxidation. In the MOD group, the NCOA4 and GPX4 proteins were upregulated and downregulated, respectively, and the latter became inactivated altogether (Figures 1 E–G). We then verified the foregoing findings by measuring the NCOA4 and GPX4 fluorescence intensities in myocardial tissue and noted that their trends were consistent with those obtained by Western blot (Figures 1 H–K). As ferritinophagy is a type of selective autophagy, we examined its association with myocardial injury. We measured the fluorescence intensities of NCOA4 and the autophagy signature indicator LC3B in the HF model. As both of these genes co-localized, their fluorescence intensities were elevated (Figures 1 L–N). The foregoing results suggest that ferritinophagy and ferroptosis are implicated in the myocardial injury that occurs in rats with post-MI HF. Ferritinophagy activated by NCOA4 upregulation and ferroptosis induced by GPX4 downregulation may have contributed to the development of myocardial injury in this model.
Figure 1
Ferritinophagy and ferroptosis are associated with myocardial injury in rats with post-infarction heart failure (HF). A – Representative echocardiographic images, B – left ventricular ejection fraction, C – left ventricular internal diameter shortening rate. D – The indicated kits measure ck-MB levels. E – Immunofluorescence intensity of NCOA4 in myocardial tissues. **p < 0.01 F – Immunofluorescence intensity of NCOA4 in myocardial tissues. G, H – Immunofluorescence intensity of GPX4 in myocardial tissues. I–K – Western blot showed protein expression levels of GPX4 (J) and NCOA4 (K). **p < 0.01 K – Western blot showed protein expression levels of GPX4 (J) and NCOA4 (K). L–N – Immunofluorescence co-localization intensity of LC3B (M) and NCOA4 (N) in myocardial tissues. These experiments were conducted on cultured CHF rat myocardial tissue. Statistical analysis was performed using the t-test and values are expressed as mean ± SD. **p < 0.01
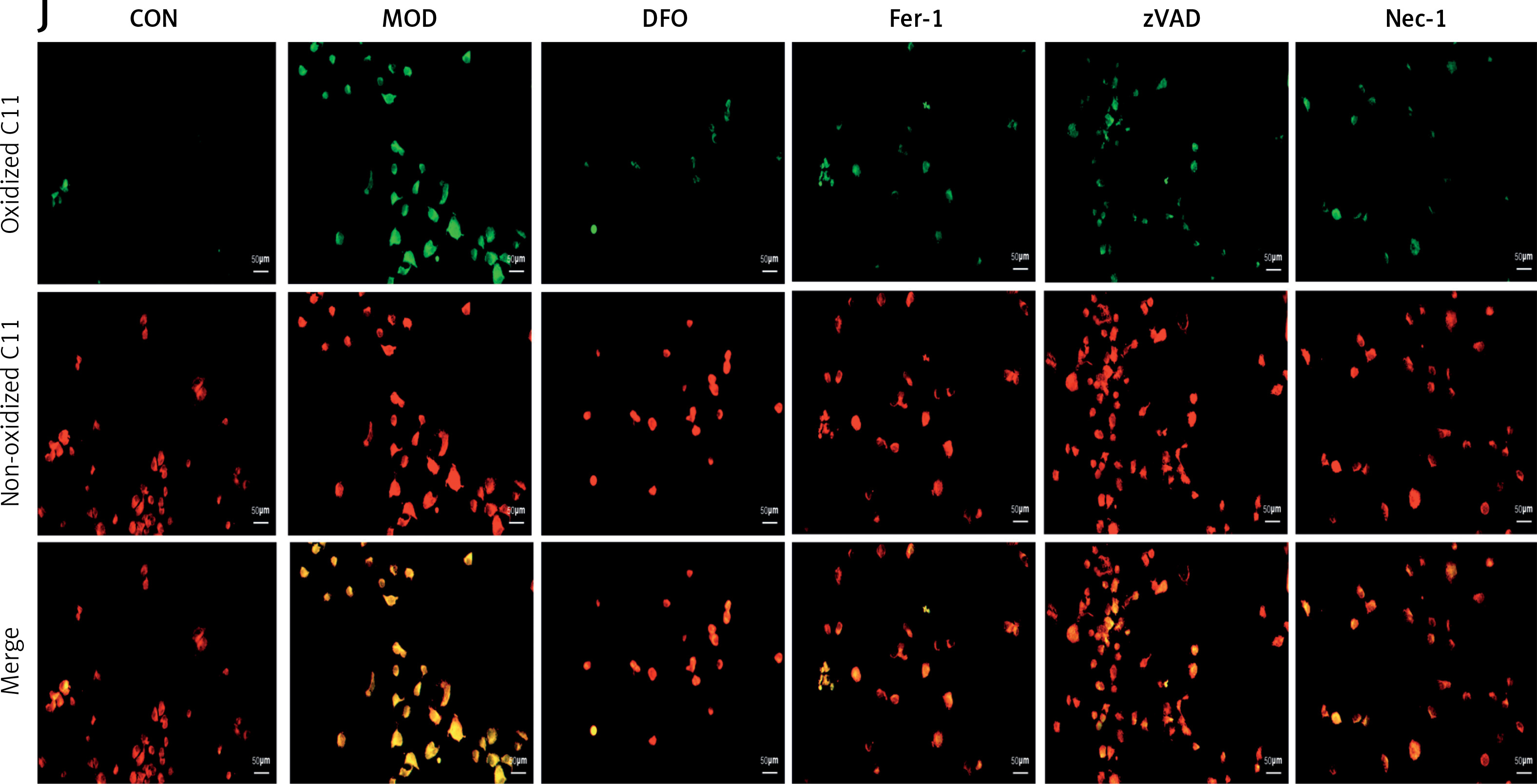
H/R induced ferritinophagy and ferroptosis in H9C2 cardiomyocytes
We then explored the involvement of ferroptosis in H/R-induced failure in H9C2 cardiomyocytes. A fluorescent probe revealed a significant increase in Fe2+-lysosome co-localization. Thus, iron had accumulated in the lysosomes in response to H/R modeling (Figures 2 A, B). We then assessed the survival of H/R-treated cells and evaluated the effects of various inhibitors on cell death. While the ferroptosis inhibitors DFO and Fer-1 and the apoptosis inhibitors Nec-1 and zVAD all inhibited H/R-induced cell death, the former two were more efficacious than the latter two (Figure 2 C). Western blot revealed that DFO and Fer-1 downregulated the key ferritinophagy protein NCOA4 and upregulated the key ferroptosis protein GPX4 (Figures 2 D–F). We then examined the impact of DFO and Fer-1 on cellular redox status. Both ferroptosis inhibitors upregulated the lipid peroxidation antagonist GSH in H/R-treated cells (Figure 2 G). They also downregulated the lipid peroxidation by-product MDA (Figure 2 H). We then used C11 BODIPY staining to assess H/R-induced lipid peroxidation and discovered that H/R exposure substantially increased lipid peroxidation. However, DFO and Fer-1 significantly reduced H/R-induced lipid peroxidation (Figures 2 I, J). We also evaluated the effects of the apoptosis inhibitors Nec-1 and zVAD on H/R-induced cardiomyocyte failure. As both compounds ameliorated H9C2 failure to a certain extent, H/R induced apoptosis as well as ferroptosis. However, the ferroptosis inhibitors were more efficacious than the apoptosis inhibitors. The foregoing findings suggest that ferritinophagy and ferroptosis may be the principal mechanisms underlying H/R-induced cardiomyocyte failure. The accumulation of iron in the lysosomes, modulation of the cellular redox status, the downregulation of NCOA4, and the upregulation of GPX4 accounted for the protective effects of the ferroptosis inhibitors in this model. Though apoptosis might also be implicated in H/R-induced cardiomyocyte failure, its overall impact on it is somewhat less than that of ferroptosis.
Figure 2
H/R induces ferritinophagy and ferroptosis in H9C2 cardiomyocytes. A, B – RhoNox (red) and LysoTracker (green) are shown by confocal imaging in H9C2 cells stimulated with H/R. These images show the fluorescence intensity and co-localization of ferrous iron and lysosomes in surviving cells. C – cell survival under induction of DFO and Fer-1 (ferroptosis inhibitors), Nec-1 and VAD (apoptosis inhibitors). D–F – Western blot showing protein expression levels of NCOA4 (E) and GPX4 (F). *p < 0.05, **p < 0.01. NS – not significant, p > 0.05 D–F – Western blot showing protein expression levels of NCOA4 (E) and GPX4 (F). G – GSH, H – MDA levels. The degree of cellular lipid peroxidation (J) was analyzed and quantified (I) using BODIPY 581/591 for the six H/R treatment groups. These experiments were conducted on cultured H9C2 myocardial cells. Data were processed by one-way ANOVA and values are expressed as mean ± SD. *p < 0.05, **p < 0.01. NS – not significant, p > 0.05 The degree of cellular lipid peroxidation (J) was analyzed and quantified (I) using BODIPY 581/591 for the six H/R treatment groups. These experiments were conducted on cultured H9C2 myocardial cells. Data were processed by one-way ANOVA and values are expressed as mean ± SD. *p < 0.05, **p < 0.01. NS – not significant, p > 0.05
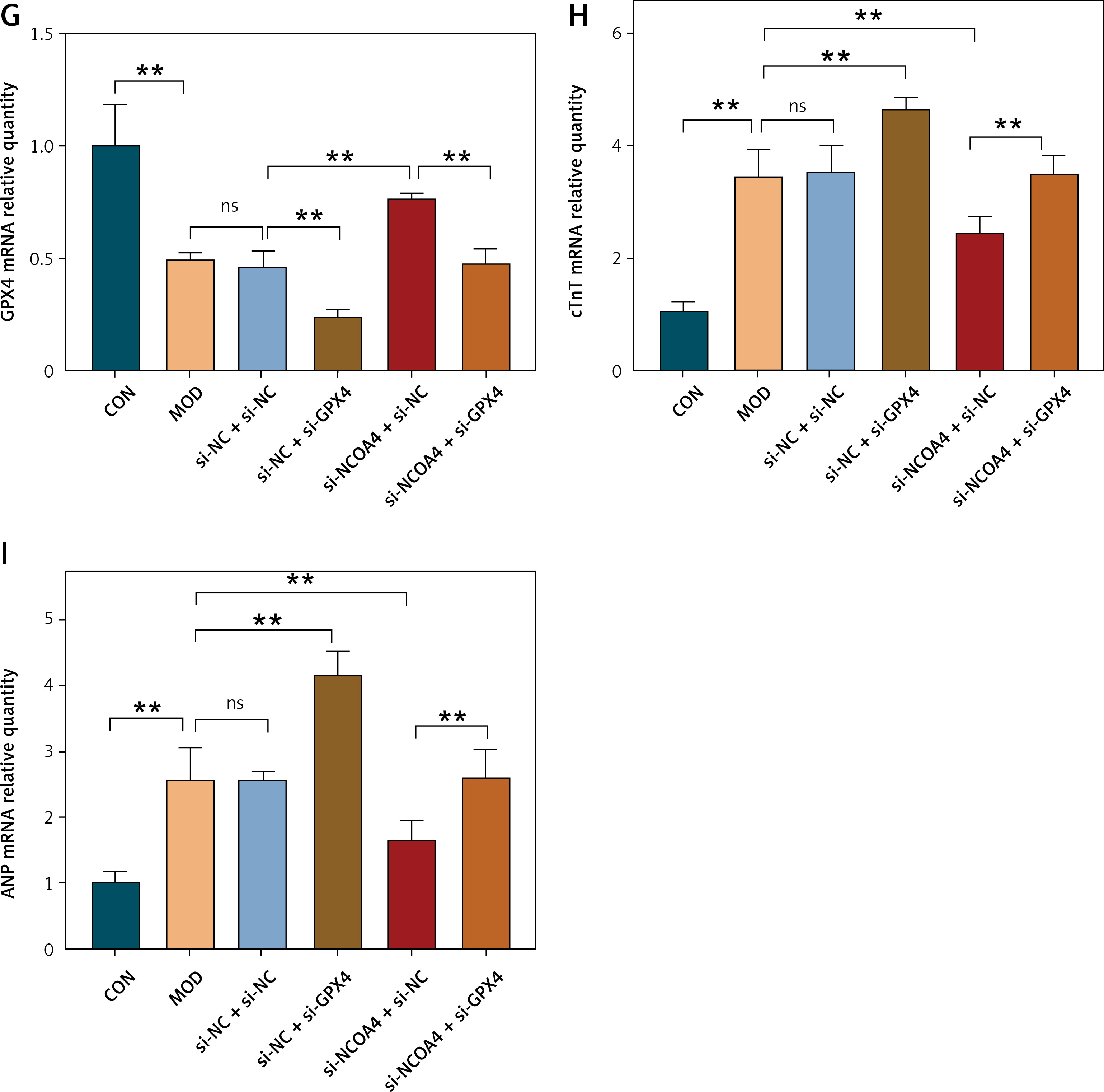
Silencing NCOA4 upregulates GPX4 and alleviates cardiac injury
The results of the foregoing experiments suggest that NCOA4-mediated ferritinophagy and GPX4-mediated ferroptosis were implicated in both the in vivo and in vitro post-MI HF models. Lipid peroxidation damages cell membranes, lipoproteins, and other lipid-containing structures, impairs normal cellular functions, and induces HF [28]. Therefore, we examined the regulatory relationship between NCOA4 and GPX4 and their effects on HF-related indices. We constructed three siRNAs targeting NCOA4 and GPX4, evaluated their efficiency by RT-qPCR, and selected si-NCOA4-1 and si-GPX4-2 (Figures 3 A, B). Western blot revealed that silencing GPX4 significantly decreased its protein expression level (Figures 3 C, D). In contrast, silencing NCOA4 significantly increased its protein expression level (Figures 3 C, D). RT-qPCR revealed the same trend at the mRNA expression level (Figure 3 G). We measured the expression levels of cTnT and ANP, which are markers of myocardial injury and cardiac dysfunction, respectively [29, 30]. Silencing GPX4 significantly upregulated both cTnT and ANP while silencing NCOA4 significantly downregulated them (Figures 3 E, F). Thus, modulating NCOA4 and GPX4 expression may impact cardiac function. We then performed a rescue experiment to confirm the relationship between NCOA4 and GPX4. Silencing NCOA4 partially attenuated the effect of GPX4 silencing and alleviated cardiac injury (Figure 3). Taken together, the preceding findings indicate that silencing NCOA4 upregulates GPX4, inhibits ferroptosis, and reduces the burden and damage on the heart. The regulatory relationship between NCOA4 and GPX4 may play a vital role in cardiac dysfunction pathogenesis and serve as a therapeutic target for HF.
Figure 3
Silencing NCOA4 upregulates GPX4 expression to alleviate cardiac injury. A, B – Expression levels of nuclear receptor cofactor 4 (NCOA4) and glutathione peroxidase 4 (GPX4) in H9C2 cells transfected with control short interfering RNA (siRNA) were measured using RT-qPCR. si-NCOA4-1 and si-GPX4-2, which have the highest silencing efficiency, were used according to the protocol. C–F – Western blot showing protein expression levels of GPX4 (D), cTnT (E), and ANP (F). **p < 0.01. NS – not significant, p > 0.05 G–I – RT-qPCR for GPX4 mRNA (G), cTnT mRNA (H), and ANP mRNA (I) is consistent with protein trends. These experiments were conducted on cultured H9C2 myocardial cells. Data were processed by one-way ANOVA and values are expressed as mean ± SD. **p < 0.01. NS – not significant, p > 0.05
Autophagy promotes NCOA4-mediated ferroptosis activation in HF
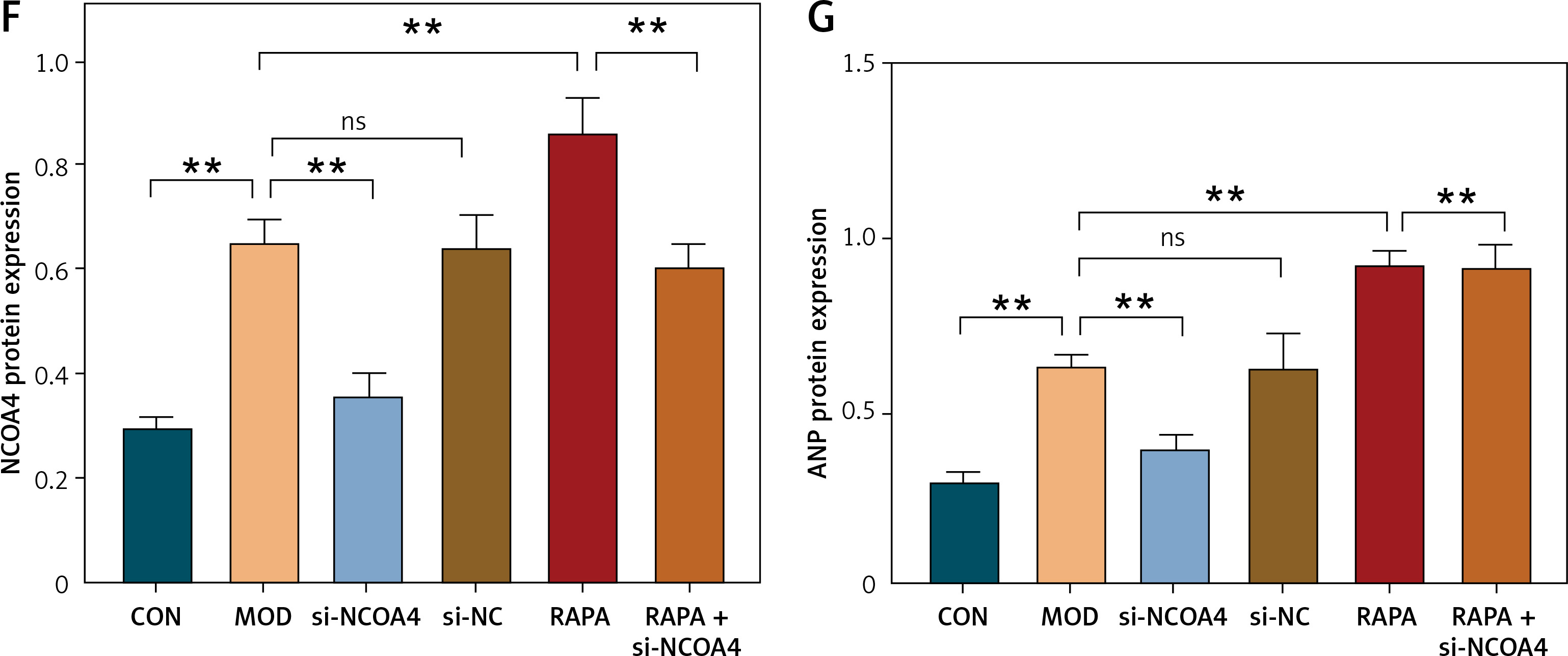
We investigated the effect of NCOA4 silencing on autophagy, intracellular iron levels, ROS production, lipid peroxidation, and NCOA4 and ANP expression following exposure to the autophagy agonist RAPA. The FeRhoNox-1 fluorescent probe showed that RAPA significantly increased the intracellular free iron content while silencing NCOA4 lowered the RAPA-induced LIP content (Figures 4 A, C). Cytoplasmic ROS production and hydroxyl radical-mediated lipid peroxidation lead to ferroptosis [31]. Here, we used flow cytometry with DCFH-DA and C11 BODIPY 581/591 fluorescent probes to measure cytoplasmic ROS intensity and lipid peroxidation intensity, respectively, and assess ferroptosis-related events. As RAPA increased the cytoplasmic/lipid ROS ratios, it increased ferroptosis intensity. However, silencing NCOA4 lowered the RAPA-induced cytoplasmic/lipid ROS ratios. Therefore, it inhibited ferroptosis (Figures 4 B, D). Western blot demonstrated that RAPA increased the protein expression of NCOA4 and ANP whereas silencing NCOA4 significantly counteracted these effects (Figures 4 O–Q). RT-qPCR revealed the same trend for ANP at the mRNA expression level (Figure 4). The foregoing findings suggest that the autophagic agonist RAPA activates NCOA4, promotes ferroptosis, and increases cardiac load while silencing NCOA4 attenuates RAPA-induced ferroptosis and cardiac load. These results demonstrate the effect of activated autophagy on NCAO4-mediated ferroptosis and myocardial injury in the HF model.
Figure 4
Autophagy promotes NCOA4-mediated ferroptosis in HF. Fluorescence intensity (A) and quantification (C) of the intracellular unstable iron pool were determined by FeRhoNox-1 fluorescent probe. B – Cytoplasmic ROS intensity, C11 BODIPY 581/591 lipid peroxidation fluorescent probe intracellular lipid peroxidation intensity, (D) and cytoplasmic/lipid ROS analysis using flow cytometry with DCFH-DA fluorescent probe. E–G – Western blot showing protein expression levels of NCOA4 (F) and ANP (G). These experiments were conducted on cultured H9C2 myocardial cells. Data were processed by one-way ANOVA and values are expressed as mean ± SD. **p < 0.01. NS – not significant, p > 0.05 E–G – Western blot showing protein expression levels of NCOA4 (F) and ANP (G). These experiments were conducted on cultured H9C2 myocardial cells. Data were processed by one-way ANOVA and values are expressed as mean ± SD. **p < 0.01. NS – not significant, p > 0.05
Ferritinophagy promotion aggravated myocardial injury in rats with post-MI HF
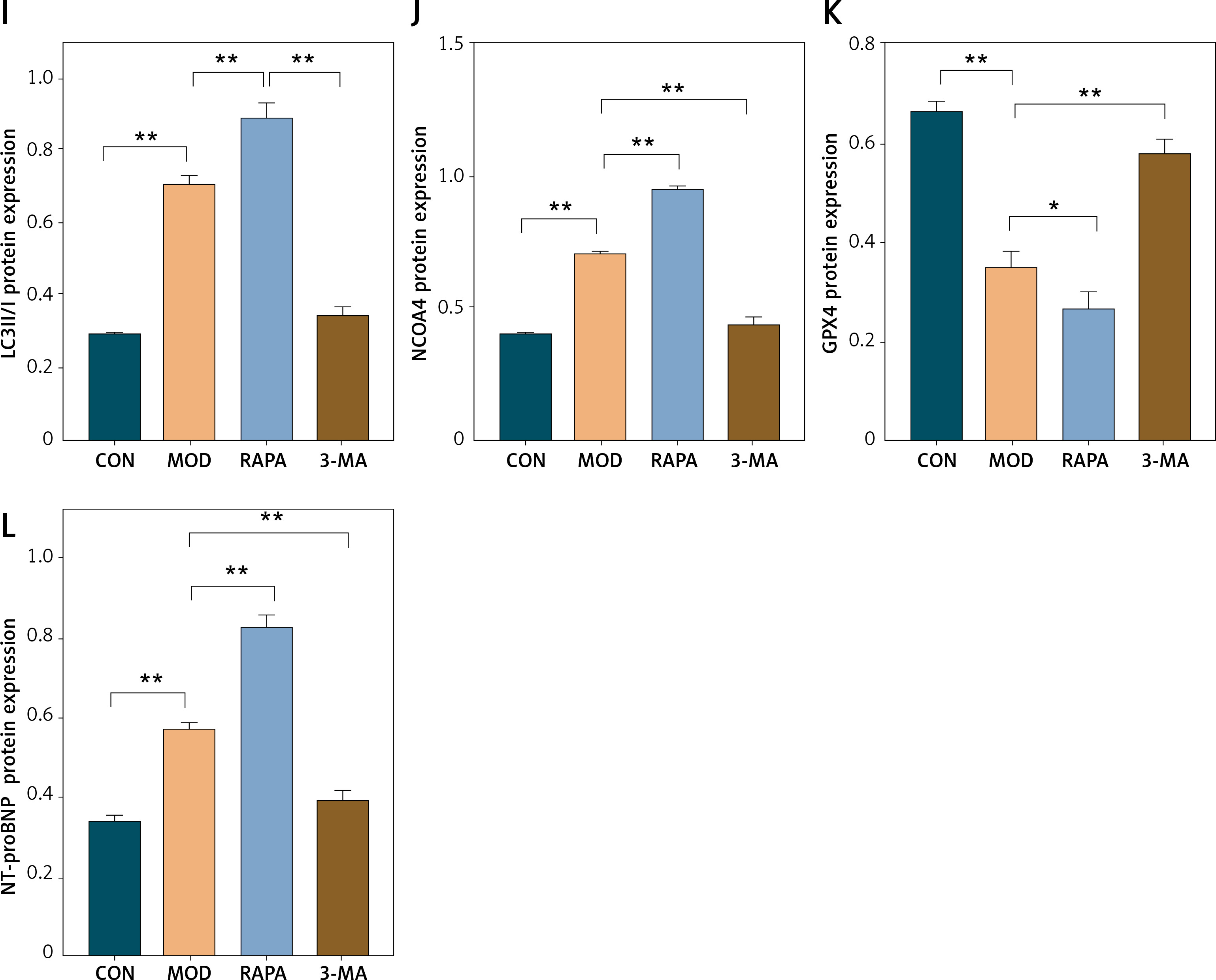
Here, we investigated the role of ferritinophagy in a rat post-MI HF model. We applied H&E staining to examine myocardial injury in the rat post-MI HF model. In the CON group, the myocardial cells were uniformly arranged and had clear borders. In the MOD group, the myocardial cells were loose, congested, swollen, and infiltrated with inflammatory cells. In the RAPA group, the myocardial cells exhibited broken myocardial fibers, coagulated and fragmented nuclei, and dense inflammatory cell infiltration. In the 3-MA group, the myocardial cells presented neatly arranged myocardial fibers, normal morphology, and minimal inflammatory cell infiltration (Figure 5 A). RAPA significantly increased the myocardial Fe2+ and Fe3+ content while 3-MA had the opposite effect (Figures 5 B, C). RAPA significantly increased MDA levels and decreased GSH and SOD in myocardial tissue, whereas 3-MA had the opposite effects (Figures 5 D–F). Under an electron microscope, the myocardial cells of the CON group exhibited evenly distributed, dense mitochondria with clear cristae. In contrast, the myocardial cells of the MOD group had disorganized, damaged, swollen, degenerating mitochondria with broken cristae and autophagic vesicles. In the 3-MA group, the mitochondria displayed intact membranes and relatively little damage. Though they were swollen and their cristae were fractured, their myocardial fibers were neatly arranged (Figure 5 G). Western blot showed that RAPA significantly upregulated the autophagy target LC3II/I and the ferritinophagy target NCOA4 but downregulated the ferroptosis target GPX4 in myocardial tissue. Conversely, 3-MA downregulated myocardial LC3II/I and NCOA4 but upregulated myocardial GPX4 (Figures 5 D–F). RAPA also upregulated the HF marker NT-proBNP, thereby revealing exacerbation of HF. In contrast, 3-MA downregulated NT-proBNP, which indicates the protection of HF. Overall, the results of the aforementioned in vivo experiments validated those of the in vitro assays. For this reason,excessive autophagy could promote the release of intracellular free iron, inhibit GPX4, accelerate oxidative stress, promote lipid peroxidation, mediate ferroptosis, and aggravate myocardial injury in the rat post-MI HF model.
Figure 5
Promotion of ferritinophagy aggravates myocardial injury in post-infarction heart failure (HF) rats. A – H&E staining to detect the extent of myocardial injury in rats with heart failure. B–F – Designated kits to detect Fe2+ (B) Fe3+ (C), GSH (D). These experiments were conducted on cultured CHF rat myocardial tissue. Data were processed by one-way ANOVA and values are expressed as mean ± SD. *p < 0.05, **p < 0.01 Designated kits to detect Fe2+ MDA (E) and SOD (F) in myocardial tissue of rats. G – Transmission electron microscopy of myocardial mitochondria. H–L – Western blot showing protein expression levels of LC3II/I (I), NCOA4 (J), GPX4 (K), and NT-proBNP (L). These experiments were conducted on cultured CHF rat myocardial tissue. Data were processed by one-way ANOVA and values are expressed as mean ± SD. *p < 0.05, **p < 0.01 H–L – Western blot showing protein expression levels of LC3II/I (I), NCOA4 (J), GPX4 (K), and NT-proBNP (L). These experiments were conducted on cultured CHF rat myocardial tissue. Data were processed by one-way ANOVA and values are expressed as mean ± SD. *p < 0.05, **p < 0.01
Discussion
Myocardium is highly sensitive to ischemia/hypoxia. Inadequate oxygen supply can lead to cardiomyocyte dysfunction and death [32]. However, few of the currently available treatments for HF are efficacious, and this condition may cause irreversible damage. Researchers commonly use in vitro H/R cell models to simulate ischemia-reperfusion and clarify the mechanisms underlying HF. Ischemia/hypoxia triggers the onset and progression of myocardial injury, and the latter ultimately leads to HF. Numerous studies have used H/R cell models to elucidate the mechanisms associated with HF development [33–35]. Post-MI HF models have been constructed in mammals to study the associations among ferritinophagy, ferroptosis, and HF, elucidate the pathophysiological mechanisms involved, and bridge the gap between in vitro experiments and clinical (in vivo) observations.
Autophagy is a tightly regulated lysosomal degradation mechanism that plays a crucial role in cell survival, homeostasis, and function [36]. Our team’s research, along with studies conducted by others, has shown that autophagy plays a significant role in regulating cardiac homeostasis and function and is closely linked to the development of HF [21–23, 37]. Impaired or overactivated autophagy in cardiac myocytes is detrimental to the maintenance of cellular homeostasis and contributes to the progression of heart failure. Some studies have indicated that autophagy activation serves as an adaptive response in dilated cardiomyopathy (DCM) patients, with increasing autophagic levels as left ventricular remodeling progresses, suggesting that autophagic markers could potentially serve as diagnostic biomarkers for heart failure [38]. While extensive research has investigated the impact of autophagy on heart failure, the specific mechanisms by which autophagic pathways influence cardiac function remain a subject worthy of further investigation. Prior research has demonstrated the critical importance of maintaining iron homeostasis for normal cellular function. Excess iron ions can induce oxidative stress and the generation of free radicals, causing damage to cell membranes and mitochondria, thereby negatively affecting cardiac myocytes [39]. Hence, HF treatment guidelines recommend iron supplementation [40]. Our in vivo experiments also revealed a substantial increase in Fe2+ and Fe3+ levels in heart tissues of HF rats. In cell experiments, the labile iron pool (LIP) levels were significantly elevated in the MOD group. Additionally, various indicators of ferroptosis, such as GSH, MDA, SOD, ROS/lipid ROS, and GPX4, were markedly altered in the MOD group, indicating the activation of ferroptosis. Excessive Fe2+ initiates the Fenton reaction, leading to overproduction of ROS and the enlargement of peroxidation in the phospholipid bilayer of organelles. Inhibition of the antioxidant enzyme GPX4 results in oxidative stress and disruption of lipid metabolism, and ultimately leads to ferroptosis [41, 42]. Our results indicate that the use of the autophagy inducer RAPA can suppress GPX4 expression, elevate NT-proBNP, and reduce heart function. Conversely, the autophagy inhibitor 3-MA can increase GPX4 expression, lower NT-proBNP, and reduce heart failure. Therefore, we suggest that autophagy may affect cardiac function through the mediation of GPX4-associated ferroptosis.
It is worth considering how autophagy influences ferroptosis. Ferritinophagy is a selective autophagic degradation of ferritin, which can induce HF. Knocking down the key ferritinophagy regulator NCOA4 in mice can alleviate pressure overload-induced cardiac dysfunction and ferritinophagy-mediated ferroptosis. Excessive activation of autophagy or lysosomal dysfunction can lead to intracellular iron accumulation and lipid peroxidation, subsequently triggering ferritinophagy and raising lipid levels. Excess intracellular free iron and lipid peroxidation decrease GPX4 expression and promote ferroptosis, which in turn accelerates HF progression [43, 44]. Our research indicates that after silencing NCOA4, the impact of RAPA on LIP and ROS/lipid ROS levels decreases, and ANP expression is downregulated, leading to an improvement in heart function. Therefore, treatment with the iron chelator DFO and the ferroptosis inhibitor Fer-1 may reverse H/R-induced cardiomyocyte iron overload by downregulating NCOA4 and upregulating GPX4. The hallmark of ferritinophagy is NCOA4-mediated ferritin degradation and the release of excess Fe2+. Conversely, silencing NCOA4 can attenuate ferritinophagy, preventing the accumulation of excess Fe2+ in cells. Furthermore, upregulation of GPX4 can inhibit ferroptosis. Previous findings suggest that NCOA4 may act upstream of GPX4, and NCOA4-mediated ferritinophagy may influence ferroptosis by regulating GPX4.
This study demonstrates that excessive autophagy can promote NCOA4-mediated ferritinophagy, affecting the release of intracellular free iron, and downregulating GPX4, thereby inducing lipid peroxidation and ferroptosis, ultimately exacerbating heart failure. Silencing NCOA4 can inhibit ferritinophagy, improving heart function, while silencing GPX4 counteracts the beneficial effects of NCOA4, worsening the severity of heart failure. Nevertheless, this study still has shortcomings, such as not revealing a direct link between NCOA4 and GPX4, and we will conduct further studies to address this issue in the future. The results of the present work lay the foundation for the development of efficacious therapeutic modalities against post-MI HF.