Introduction
Venous thromboembolism (VTE), encompassing pulmonary embolism (PE) and deep vein thrombosis (DVT), is a serious thromboembolic disorder that remains a substantial challenge for global healthcare systems [1]. VTE is a multifactorial disease arising from the interplay between genetic and acquired risk factors, with its heritability estimated at 40% to 60% based on studies involving families, twins, and siblings [2]. A recent study indicated that the annual incidence of VTE in the USA is 123 cases per 100,000 people, with a higher rate among the elderly [3]. PE is more severe, often causing acute obstruction of the pulmonary vasculature, leading to hemodynamic instability and even death [4]. Among cardiovascular complications worldwide, DVT is the third leading cause of death and disability [5]. Therefore, developing novel strategies for the prevention and treatment of VTE is essential to mitigate the socioeconomic consequences associated with its incidence and progression.
Anticoagulation therapy is the primary treatment for VTE, and delays in initiating or maintaining therapeutic levels may lead to poorer outcomes [6]. Rivaroxaban, a non-vitamin K oral anticoagulant (NOAC), is extensively employed for patients at heightened risk of thrombosis, especially VTE [7]. Preventing VTE is a crucial goal for medical professionals, requiring a thorough understanding of the risk factors to effectively mitigate the risk. A multicenter cohort study identified the neutrophil-to-lymphocyte ratio (NLR), lactate dehydrogenase (LDH), C-reactive protein (CRP), and procalcitonin (PCT) as independent predictive factors for VTE [8]. Another Mendelian randomization (MR) analysis found that, among the 41 inflammatory cytokines included, only platelet-derived growth factor-BB (PDGF-BB) levels showed a causal relationship with an increased risk of VTE, PE, and DVT [9]. Further research is needed to identify precise risk factors for VTE.
The human gut harbors trillions of microorganisms, which play a crucial role in maintaining digestive health and immune homeostasis [10]. Research showed that alterations in the composition of these GM are associated with various diseases, including gastrointestinal disorders, metabolic issues, and cardiovascular conditions [11–13]. Environmental or genetic disturbances in GM can trigger inflammatory reactions in blood vessels, platelets, and immune cells, potentially increasing the risk of thrombosis [14]. Disruption of the intestinal epithelial barrier, caused by factors such as inflammation, nutrition, and antibiotics, allowed microbial products and metabolites to enter the systemic circulation via the portal vein, potentially leading to thrombosis [15]. Disturbances in GM can activate pathways involving endothelial cells, platelets, and innate immune cells, leading to the release of coagulation proteins and the development of a prethrombotic state [16]. However, the precise role of GM in the development of thromboembolism is not yet fully understood.
MR uses natural variations in genetic variants across generations to determine causal relationships [17]. By employing single nucleotide polymorphisms (SNPs) associated with specific health conditions as proxies, MR helps identify causal relationships while avoiding the external biases often present in traditional epidemiological studies [18–20]. This approach provides a clearer understanding of the genetic influences on disease.
Our research investigated the influence of GM on venous thromboembolic conditions, including VTE, PE, and DVT, using two-sample summary MR with genetic variants associated with GM as instruments to explore these relationships.
Material and methods
Study design
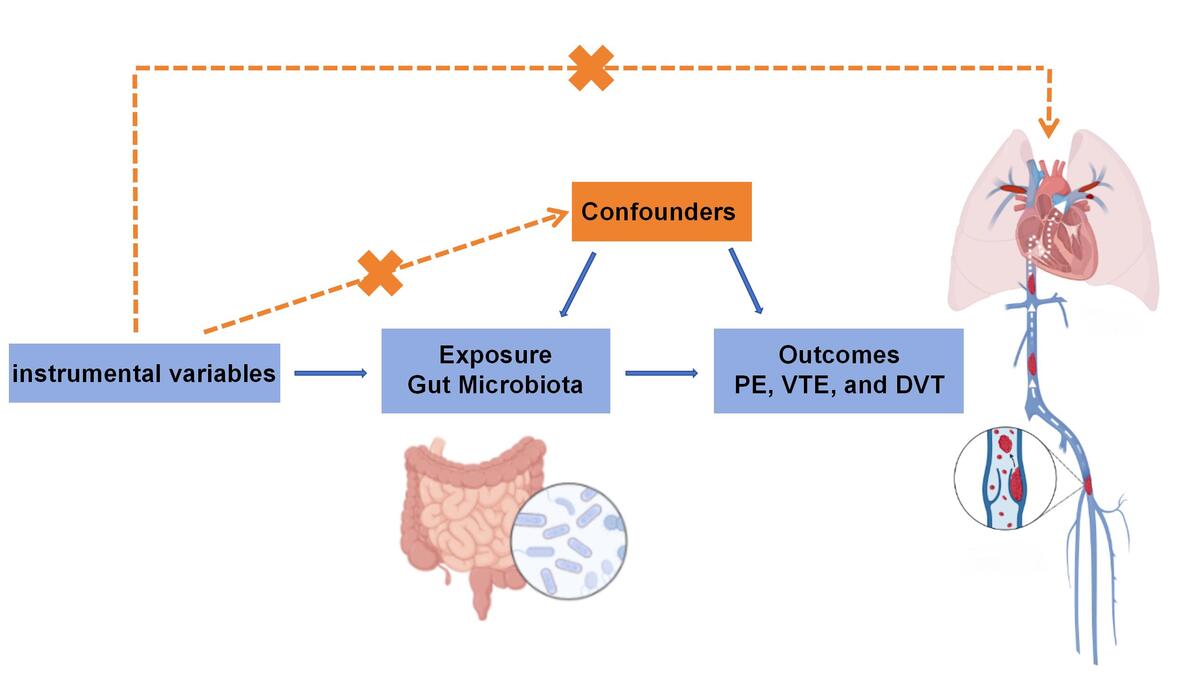
This study was designed in accordance with Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization (STROBE-MR). SNPs associated with the human GM were used as instrumental variables (IVs), with VTE, including PE and DVT, as the outcome variable. The study satisfies the three core assumptions of MR analysis: (1) the relevance assumption, indicating that the IVs are significantly associated with the exposure (GM); (2) the independence assumption, ensuring that the IVs are not associated with any confounding variables; and (3) the exclusion restriction assumption, which states that the selected genetic variants influence the outcome exclusively through the “IVs-exposure-outcome” pathway, without affecting the outcome via alternative pathways (Figure 1). The data for this study were aggregated from previously published research, for which participant consent and ethical clearance had been obtained.
Data resources
Forward MR data
The genetic data related to GM were obtained from the global MiBioGen consortium database (https://mibiogen.gcc.rug.nl/), which integrates data from 25 cohorts across multiple countries, including the USA and Italy, involving a total of 18,340 individuals. The primary objective of this study was to identify the relationship between autosomal human genetic variants and GM by analyzing the participants’ 16S rRNA sequencing profiles [21]. The genome-wide association study (GWAS) data for VTE, PE, and DVT were derived from the FinnGen consortium R9 release dataset. This genetic dataset includes 19,372 VTE cases and 357,905 controls, 9,109 9,243 PE cases and 367,108 controls, as well as DVT cases and 324,121 controls (Table I).
Reverse MR data
For reverse MR, we used data similar to those of the forward MR approach. In this context, VTE, PE, and DVT are considered variables of interest, and SNPs strongly associated with these conditions (p < 5 × 10–8) are identified as exposure variables. This process involves removing instances of linkage disequilibrium, palindromic sequences, and weakly correlated variables, as well as SNPs influenced by confounding factors, just as in forward MR.
IV selection
Based on the three core assumptions of MR analysis, we first selected SNPs significantly associated with GM as IVs. SNPs with P-values less than the genome-wide significance threshold (p < 5 × 10–8) were chosen as the initial IVs. Secondly, to eliminate linkage disequilibrium (LD), SNPs within an LD region defined by a distance of 10,000 kb and an LD r2 < 0.001 were excluded. Subsequently, SNPs associated with known confounding factors, such as cancer, prolonged bed rest, and fractures [22], were removed (http://www.phenoscanner.medschl.cam.ac.uk/). Finally, to ensure allele alignment accuracy, harmonization of SNPs was performed [23]. The F-statistic was calculated using the formula: F = R2(N – K – 1)/[K(1 – R2)], where R2 represents the proportion of variance explained by the SNPs, N is the number of participants in the exposure group, and K is the total number of SNPs included in the final analysis. An F-statistic ranging from 26.604 to 26.992 indicates a low risk of weak instrument bias. These screening steps ensure the robustness and reliability of the study results.
MR analysis
In this study, the inverse variance weighted (IVW) method was employed as the primary analytical approach. In the forward MR analysis, GM served as the exposure variable, with SNPs associated with GM used as IVs to evaluate the causal relationships between VTE, PE, and DVT. Cochran’s Q test was conducted to assess heterogeneity across genetic instruments. When p < 0.05, the IVW random-effects model was applied to estimate the causal effect; when the p ≥ 0.05, a fixed-effects model was used [24]. To further validate and complement the causal inference results, additional methods were applied, including MR-Egger regression, weighted median estimator (WME), simple mode (SM), and weighted mode (WM) approaches. The MR-Egger method incorporates an intercept into the regression model to detect and adjust for horizontal pleiotropy in IVs, thereby enhancing the robustness of causal effect estimates [25]. The WME method calculates the weighted median of IV effects, thereby reducing the influence of outliers or biased IVs on the overall results [26]. The SM method estimates the mode of the causal effect distribution, providing reliable estimates when most IVs exhibit similar effects [27]. The WM method calculates the weighted mode of effect results, offering more robust estimates in the presence of effect heterogeneity, particularly when data points have different weights or when outliers are present [28]. By integrating these methods, this study aimed to enhance the accuracy and robustness of causal effect estimates.
Statistical analysis
In this study, sensitivity analyses included Cochran’s Q test, the MR-Egger intercept test, the MR-PRESSO method, and leave-one-out analysis. Cochran’s Q test was performed to assess the heterogeneity of the effects of the included IVs or SNPs, aiming to determine whether significant differences exist between data and estimates from different sources. P < 0.05 indicates significant heterogeneity, in which case the IVW random-effects model should be applied; if no significant heterogeneity is detected, the IVW fixed-effects model is used. The MR-Egger intercept test was employed to evaluate and quantify the horizontal pleiotropy of the instrumental variables, aiming to detect and correct potential bias in causal effect estimates. The MR-PRESSO method improves the accuracy of causal inference by identifying and removing outlier SNPs, thereby detecting and adjusting for horizontal pleiotropy. The leave-one-out analysis assesses the robustness and reliability of the results by sequentially removing each SNP and re-evaluating the causal estimates using the remaining SNPs. MR analyses were reported using p-values, odds ratios (ORs), and 95% confidence intervals (CIs). A significance level of α = 0.05 was applied for causal inference. β > 0 indicates a positive association between the microbiota and the disease, whereas β < 0 suggests a negative association. Similarly, an OR > 1 indicates a positive association, while an OR < 1 suggests a negative association between the microbiota and the disease. All MR analyses were performed using R software (version 4.3.1) with the “Two Sample MR” package (version 0.5.6).
Results
IV selection
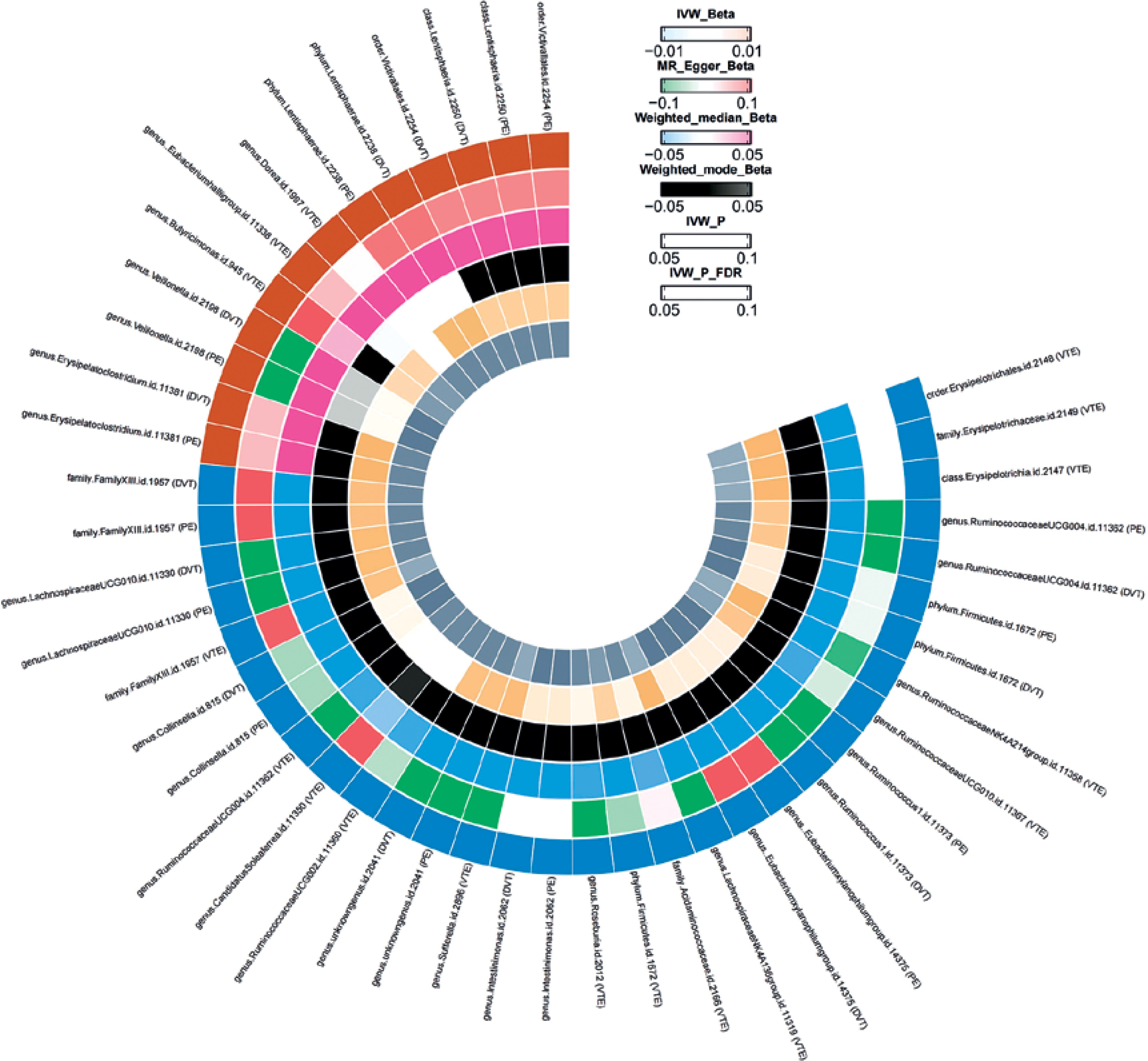
According to the selection criteria for IVs, we identified 2,779 eligible SNPs. This dataset includes 211 GM taxa, comprising 131 genera, 35 families, 20 orders, 16 classes, and 9 phyla. F-statistics were calculated for each of the 2,779 SNPs, and no SNPs with F < 10 were identified, suggesting a low likelihood of weak instrument bias affecting the causal associations. Based on the IVW analysis, we found that most of the 211 GM taxa were not associated with VTE, whereas only a few taxa showed significant associations. Moreover, different types of thrombotic diseases were associated with specific bacterial taxa (Figure 2). In total, we identified 17 SNPs causally related to VTE (Supplementary Table SI), 14 SNPs associated with PE (Supplementary Table SII), and 17 SNPs associated with DVT (Supplementary Table SIII).
Forward-direction MR analyses
A total of 14 gut bacterial taxa were found to have significant associations with VTE. Among them, Candidatus Soleaferrea (id.11350, OR = 0.92, 95% CI: 0.85–1.00, p = 0.047), Ruminococcaceae UCG002 (id.11360, OR = 0.92, 95% CI: 0.85–1.00, p = 0.046), and Ruminococcaceae UCG004 (id.11362, OR = 0.92, 95% CI: 0.85–1.00, p = 0.046) were negatively associated with VTE, while the Eubacterium hallii group (id.11338, OR = 1.12, 95% CI: 1.01–1.23, p = 0.025), Butyricimonas (id.945, OR = 1.11, 95% CI: 1.01–1.22, p = 0.027), and Dorea (id.1997, OR = 1.14, 95% CI: 1.00–1.31, p = 0.047) were positively associated with VTE.
A total of 9 gut bacterial taxa were found to have significant associations with PE. Among them, Intestinimonas (id.2062, OR = 0.88, 95% CI: 0.78–0.99, p = 0.036), an unknown genus (id.2041, OR = 0.87, 95% CI: 0.77–0.97, p = 0.015), and Firmicutes (id.1672, OR = 0.86, 95% CI: 0.74–0.99, p = 0.036) were negatively associated with PE, while Veillonella (id.2198, OR = 1.22, 95% CI: 1.01–1.48, p = 0.043), Erysipelatoclostridium (id.11381, OR = 1.16, 95% CI: 1.04–1.30, p = 0.007), and Lentisphaerae (id.2250, OR = 1.14, 95% CI: 1.03–1.26, p = 0.023) were positively associated with PE.
A total of 13 gut bacterial taxa were found to have significant associations with DVT. Among them, Mollicutes (id.3920, OR = 0.96, 95% CI: 0.84–1.09, p = 0.004), Actinobacteria (id.419, OR = 0.85, 95% CI: 0.74–0.97, p = 0.017), and Bifidobacteriaceae (id.433, OR = 0.85, 95% CI: 0.74–0.99, p = 0.031) were negatively associated with the disease, while Adlercreutzia (id.812, OR = 1.80, 95% CI: 0.93–3.50, p = 0.046), Collinsella (id.815, OR = 1.31, 95% CI: 1.08–1.59, p = 0.006), and Desulfovibrio (id.3173, OR = 1.16, 95% CI: 1.01–1.33, p = 0.006) were positively associated with the disease (Table II).
Table II
MR analysis of the causal relationship between GM and VTE, PE, and DVT
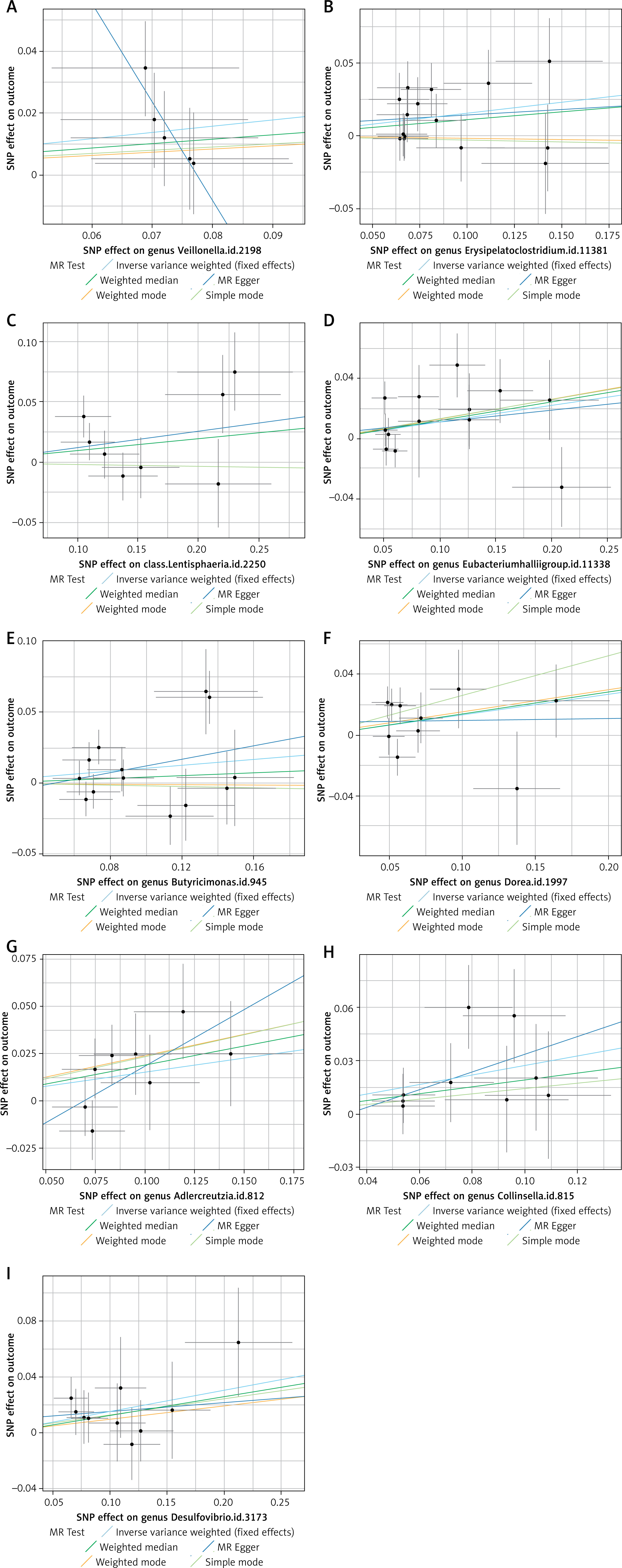
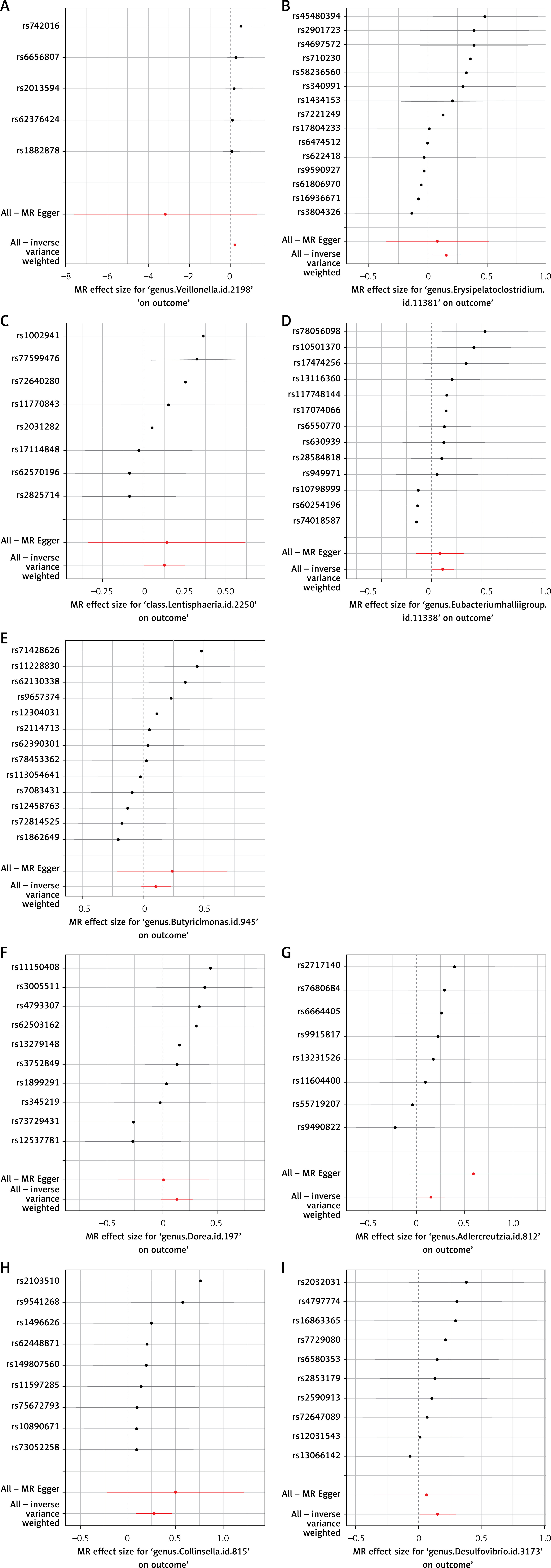
This MR analysis revealed a positive causal relationship between certain GM and the risk levels of VTE, PE, and DVT. After multiple testing correction using the Bonferroni method, a significant causal association was identified between Ruminococcaceae and VTE. This suggests that changes in GM may have corresponding effects on thrombotic diseases such as VTE, PE, and DVT. Additionally, the results of supplementary analyses, including MR-Egger, WME, and ML methods, were consistent with the direction of the IVW method. The statistically significant associations are illustrated by scatter plots and forest plots (Figures 3 and 4).
Reverse-direction MR analyses
In the reverse MR analysis, we applied the same analytical procedures, setting GM as the outcome and VTE, PE, and DVT as the exposure factors. The results showed no causal relationships between GM and VTE (p > 0.05), PE (p > 0.05), or DVT (p > 0.05) (Supplementary Tables SIV–SVI).
Sensitivity analysis
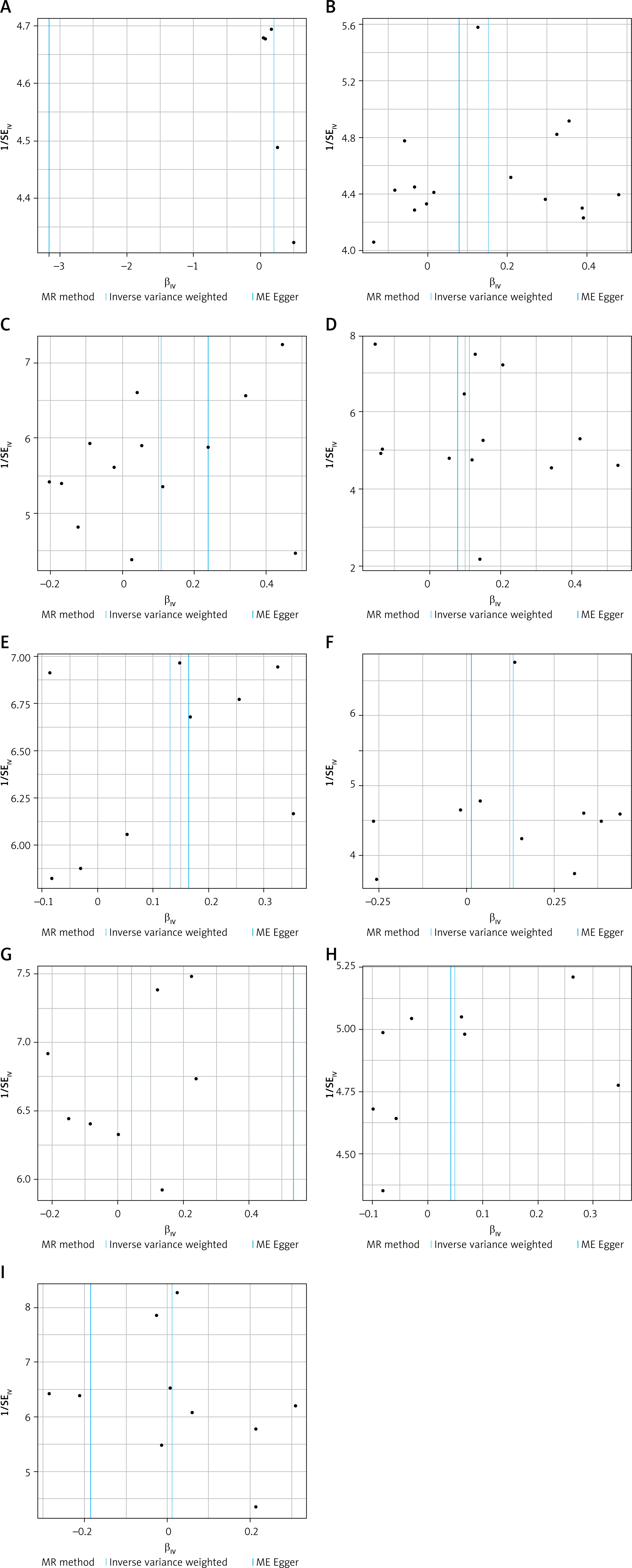
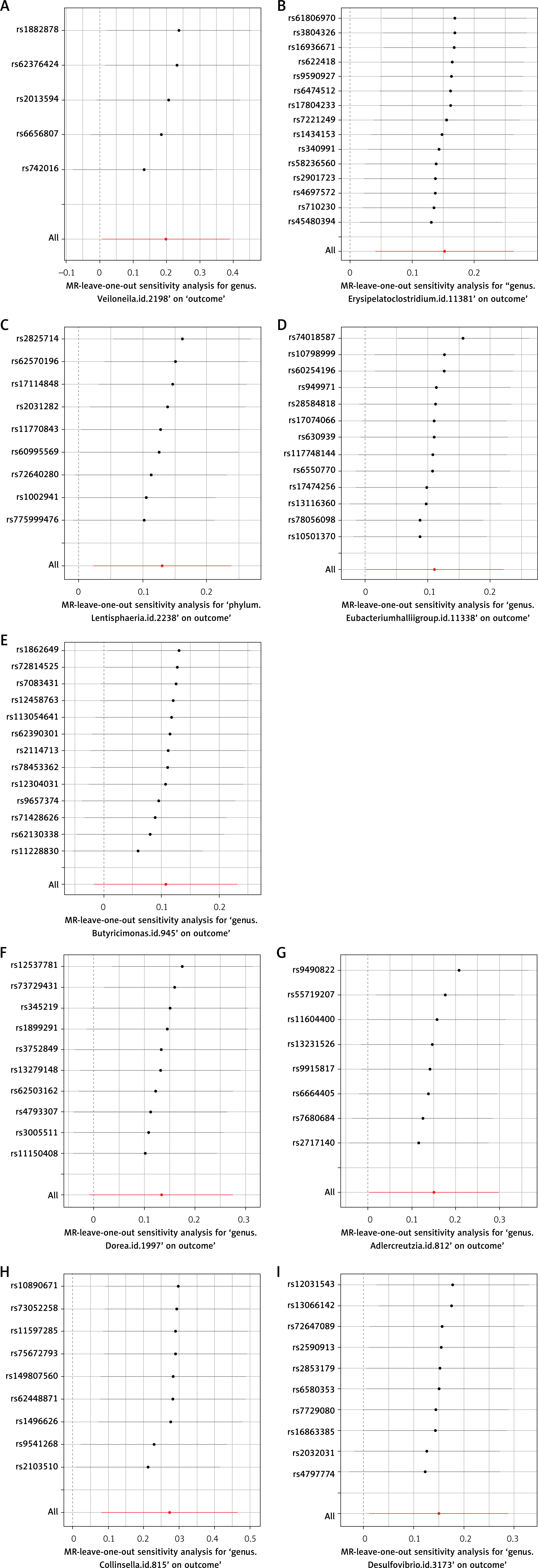
The funnel plot generated from the MR-Egger regression (Figure 5) indicated no evidence of heterogeneity or horizontal pleiotropy in the forward MR results. Additionally, no outliers were detected in the MR-PRESSO analysis or Cochran’s Q test. A leave-one-out analysis was subsequently performed, and the corresponding forest plot (Figure 6) further confirmed the stability of these results.
Discussion
To gain a more comprehensive understanding of the impact of GM on VTE development, this study conducted bidirectional two-sample MR analyses using summary statistics for VTE, PE, and DVT from the FinnGen Consortium R9 data, along with GM meta-analysis data from the global MiBioGen Consortium. A total of 211 GM taxa were analyzed for causal associations with VTE, PE, and DVT. The analysis identified causal associations between 18 GM taxa and the incidence of VTE, PE, and DVT. Specifically, Candidatus Soleaferrea, Ruminococcaceae UCG002, and Ruminococcaceae UCG004 were negatively associated with VTE, while Eubacterium hallii group, Butyricimonas, and Dorea showed positive associations with VTE (p < 0.05 and OR > 1). For PE risk, Intestinimonas, an unknown genus, and Firmicutes were negatively associated, whereas Veillonella, Erysipelatoclostridium, and Lentisphaerae showed positive associations (p < 0.05 and OR > 1). Regarding DVT risk, Mollicutes, Actinobacteria, and Bifidobacteriaceae were negatively associated, while Adlercreutzia, Collinsella, and Desulfovibrio were positively associated (p < 0.05 and OR > 1). These findings enhance the understanding of GM’s role in VTE pathogenesis and highlight specific taxa that may contribute to or protect against VTE progression.
The pathogenesis of VTE is highly complex. Thrombosis is a pathological process characterized by the abnormal aggregation and solidification of blood components within blood vessels, leading to vascular obstruction, driven by endothelial injury, altered hemodynamics, and a hypercoagulable state [29]. While thrombosis is essential for hemostasis in damaged vessels, it can also result in adverse events such as vascular occlusion, embolism, and pathological clot formation. Conversely, impaired thrombosis may lead to excessive bleeding [30]. The regulatory role of GM in VTE development has increasingly become a research focus, with different bacterial taxa potentially influencing VTE risk through metabolic products, inflammation modulation, and gut barrier function. Ruminococcaceae is a key GM family closely associated with various metabolic processes and diseases. Although direct studies on the association between Ruminococcaceae and VTE are limited, metabolites produced by GM, particularly the metabolite trimethylamine-N-oxide (TMAO), have been shown to be associated with VTE [31]. One study reported that, compared to low TMAO levels, patients with moderate and high TMAO had a 38% and 44% increased risk of VTE recurrence, respectively. However, the results were not statistically significant [32]. Papa et al. reported that TMAO is a risk factor for inflammatory bowel disease, with Ruminococcaceae playing a crucial role in its production and potentially influencing the thrombotic process [33]. Ruminococcaceae metabolize dietary lipids, including choline, phosphatidylcholine, and L-alpha glyceryl phosphorylcholine, into trimethylamine (TMA), which is subsequently oxidized to TMAO in the liver. Elevated TMAO levels have been linked to endothelial dysfunction, platelet hyperreactivity, and increased thrombosis risk [34]. Notably, Ruminococcaceae abundance is negatively correlated with thrombosis formation, suggesting that a higher abundance of Ruminococcaceae may be associated with a lower risk of VTE.
Additionally, a study by Huang et al. involving 33 patients with liver cirrhosis found a positive correlation between Eubacterium hallii group and the occurrence of VTE. This bacterial group was significantly enriched in patients with both liver cirrhosis and VTE, and its abundance was positively associated with coagulation factor parameters [35]. These findings align with the results of our study. Butyricimonas is a GM primarily known for producing butyrate, a key short-chain fatty acid with anti-inflammatory properties that plays a crucial role in maintaining intestinal barrier function. Disruption of inflammation and intestinal barrier integrity has been identified as a potential trigger for VTE [36]. Therefore, it has been hypothesized that Butyricimonas may indirectly influence VTE risk by modulating inflammation or preserving gut health. A reduction in Butyricimonas abundance could theoretically promote thrombosis through inflammatory pathways. However, our findings contradict this assumption, suggesting that the association between Butyricimonas and VTE may involve more complex underlying mechanisms. Dorea belongs to the Firmicutes phylum and is frequently associated with metabolic diseases such as obesity and diabetes, as well as inflammatory conditions, all of which are important risk factors for VTE [37]. In cardiovascular disease-related studies, changes in Dorea abundance have been linked to inflammation and metabolic dysregulation, potentially influencing VTE risk by activating coagulation pathways or increasing blood viscosity [38]. If an increase in Dorea abundance is associated with exacerbated inflammation or metabolic disturbances, it may indirectly elevate the risk of VTE through these mechanisms. However, Dorea may also contribute to reducing VTE risk by maintaining GM stability or supporting anti-inflammatory effects. Therefore, the precise association between Dorea and VTE (whether positive or negative) requires further investigation and validation. Currently, no clear research has explored the association between Candidatus Soleaferrea and VTE risk, highlighting a potential direction for future studies.
The GM may play a crucial role in the occurrence and progression of PE through the regulation of the “gut-lung axis.” Wu et al. demonstrated that certain drugs could alleviate pulmonary inflammation in mice by modulating Intestinimonas, and this inflammatory response is closely associated with thrombosis risk. It has been hypothesized that an increase in Intestinimonas abundance may exert a protective effect against PE [39]. Further research has revealed that Intestinimonas influences pulmonary inflammation and immune responses through the “gut-lung axis”, contributing to immune homeostasis in the lungs [40]. Conversely, a decrease in Intestinimonas abundance may exacerbate systemic inflammation, thereby increasing the risk of PE. Our findings are consistent with this evidence. Veillonella is an anaerobic bacterium that may play a crucial role in PE formation. A case report described a 38-year-old female patient who developed Lemierre’s syndrome following a throat infection, with imaging revealing thrombosis in the jugular and subclavian veins, accompanied by systemic complications. Blood culture identified Veillonella parvula, suggesting that this bacterium may contribute to the formation of infectious thrombosis [41]. This finding also supports our study conclusions. Additionally, recent studies have found that the GM composition in patients with idiopathic pulmonary arterial hypertension differs from that of healthy controls, with Firmicutes exhibiting the highest abundance at 53.16% and 57.08%, respectively [42]. Another study demonstrated that cryptotanshinone alleviates pulmonary fibrosis by modulating GM and bile acid metabolism, significantly reducing the proportion of Erysipelatoclostridium, suggesting its potential involvement in pulmonary diseases [43]. However, the specific association between Erysipelatoclostridium and PE requires further investigation. Regarding the unknown genus and Lentisphaerae, there is a lack of research on their association with PE in the existing literature, and no direct evidence currently supports their association with PE.
Research on the role of GM in DVT remains in its early stages. A metabolomics study identified altered metabolic profiles in DVT patients, suggesting that GM may contribute to DVT pathogenesis [44]. Studies indicated that Collinsella is enriched in DVT patients with myelofibrosis, producing short-chain fatty acids and other metabolites that influence host metabolism and immune function [45]. This underscores the significance of GM in DVT progression and its potential as a diagnostic and therapeutic target. However, no studies have directly reported associations between Mollicutes, Actinobacteria, Bifidobacteriaceae, Adlercreutzia, Desulfovibrio, and DVT.
With aging, GM diversity increases, and its composition and function tend to stabilize. The dominant bacterial phyla in the adult gut include Bacteroidetes, Firmicutes, Actinobacteria, and Proteobacteria. These active microbial communities play a crucial role in carbohydrate metabolism, energy production, cell component synthesis, nutrient processing, and immune system development [46]. However, the vast diversity of GM poses significant challenges for comprehensive measurement and quality control. Additionally, the effects of the same GM taxa may vary across different diseases. Therefore, this study employs MR analysis to investigate the association between GM and the risk of VTE, PE, and DVT. The strategy of targeting the GM as a host regulatory factor has gradually attracted attention in emerging therapeutic approaches for chronic diseases. These approaches include fecal microbiota transplantation, probiotic supplementation, dietary interventions, targeted use of antibiotics, and inhibition of specific microbial enzymes [47–51]. GM is also closely linked to intestinal inflammation. However, most supporting evidence for these associations is indirect, necessitating further direct experiments, modeling studies, and comprehensive investigations for validation.
Our study offers several advantages. It is among the few MR analyses that have investigated the causal association between GM and VTE. We incorporated extensive GWAS data from multiple databases, ensuring high-quality instrumental variables with F > 10, thereby reducing the risk of weak instrumental bias and enhancing explanatory power. However, certain limitations should be acknowledged. The GWAS data primarily involve individuals of European descent, which may restrict the generalizability of our findings to other ethnic groups. Additionally, most GWAS studies employ 16S rRNA gene sequencing at the genus level, preventing precise association of specific strains or species with our findings. Moreover, due to limitations in the outcomes database, the phenotypes discussed do not completely encompass all types of VTE, PE, and DVT, thereby somewhat reducing the clinical relevance and interpretative depth of our conclusions. Future large-scale clinical trials and cohort studies are necessary to further validate our findings.
In conclusion, specific GM exhibit a clear causal relationship with the development of VTE, PE, and DVT. Ruminococcaceae was found to significantly reduce the risk of VTE. This study enhances the understanding of the role of GM in VTE pathogenesis, particularly in the potential mechanisms of the microbiota-immune-coagulation network. Moreover, it provides important theoretical support for the development of innovative therapeutic approaches based on probiotics or microbiota transplantation.