Introduction
Lymphoma is a malignant neoplasm originating from lymphatic tissue, capable of metastasizing to any region of the body, and presenting a range of clinical manifestations [1]. The most prevalent hematologic malignancy, non-Hodgkin’s lymphoma (NHL), which accounts for 90% of all cases, and Hodgkin’s lymphoma, which accounts for 10% of all cases, are the two traditional categories for lymphomas [2]. NHL has both aggressive and inert subtypes with a 5-year overall survival rate that ranges from 25% to 75%, and the prognosis is variable [3]. Genetic factors play a 10% role in the onset of lymphoma, and there is no significant difference in heritability between the sexes [4]. Many subtypes of lymphoma remain incurable with current management strategies, and more clinical trials are needed to determine novel therapies with promising activity in this disease [5].
The human intestinal flora is a collection of various microorganisms, including bacteria, fungi, and viruses, that live on the surface of the epithelial barrier of the gastrointestinal tract [6, 7]. With the advancement of molecular tools and technologies, such as 16S ribosomal RNA sequencing and metabolomics, complex host-microbiota interactions are gradually being discovered [8, 9]. Numerous studies have shown that gut microbiota (GM) play key roles in immune, metabolic and inflammatory responses [10, 11].
Studies have shown that the combination of gut microbiota and vitamin D is promising for the treatment and prevention of autoimmune and allergic diseases and that Th17 cells also play a key role [12, 13].
In addition, it has been suggested that dysregulation of gut microbial ecology jeopardizes the integrity of the immune network and may lead to acute lymphoblastic leukemia [14]. Among them, a group of potential tumor-promoting or anti-tumor microbial species have been identified [15], which lays the foundation for modulating the gut flora in cancer therapy.
An observational study found significant enrichment of Faecalibacterium, Bifidobacterium, and Ruminococcus abundance in patients with NHL treated with CD19 CAR-T cells [16].
Genetic variation is used as an instrumental variable (IV) in Mendelian randomization (MR), a technique that is widely used to explore potential causal relationships between environmental exposures and disease and also a widely accepted way to control for potential confounders and avoid reverse causality bias [17–19]. Combining single nucleotide polymorphism (SNP) exposure and SNP outcome associations from a separate genome-wide association study (GWAS) with MR studies of two samples allows for the creation of a single causal estimate. MR analysis must be predicated on three fundamental tenets in order to guarantee the validity of our findings: (1) genetic variants must exhibit a strong correlation with the exposure factor; (2) genetic variants must not have a direct impact on the outcome; and (3) genetic variants must not be causally linked to any potential confounders [20].
Randomized controlled trials of the gut microbiota, as opposed to observational studies, may aid in the establishment of causation. Unfortunately, due to objective elements such as technology and study technique, strain screening involving early diagnosis and prognosis still has considerable limitations. These experiments were impacted by a number of variables, including antibiotic use and diet [21, 22]. In conclusion, it is still unclear whether GM and lymphomas (Hodgkin’s and non-Hodgkin’s lymphomas) are causally related.
Material and methods
Exposure data
Summary statistics of GM from the global consortium MiBioGen’s GWAS dataset served as exposure IVs [23, 24]. This was a multi-ethnic large-scale GWAS that coordinated genome-wide genotype and 16S fecal microbiome data from 18,340 participants from 24 cohorts in the United States, Canada, Israel, Korea, Germany, Denmark, the Netherlands, Belgium, Sweden, Finland, and the United Kingdom to explore the relationship between human autosomal genetic variation and the GM. A total of 211 taxa were included, belonging to 35 families, 20 orders, 16 phyla, 9 phyla and 131 genera. The externally validated genetic data on the gut microbiota were obtained from the GWAS summary data available on the IEU Open GWAS Project website (https://gwas.mrcieu.ac.uk/).
Outcome data
GWAS summary statistics for Hodgkin’s and follicular lymphoma (HL and FL), mature T/NK-cell lymphoma (MT/NKCL), and diffuse large B-cell lymphoma (DLBCL) (all with exclusion of other cancers) were obtained from the FinnGen Consortium (updated to 2021, total Ncase = 1,250, Ncontrol = 716,274). To facilitate analysis and data extraction, we accessed and analyzed outcome data directly through the OPEN GWAS website (https://\/GWAS.mrcieu.ac.uk/). Detailed information is provided in Table I.
Table I
Overview of the source of lymphoma data
Instrumental variable selection
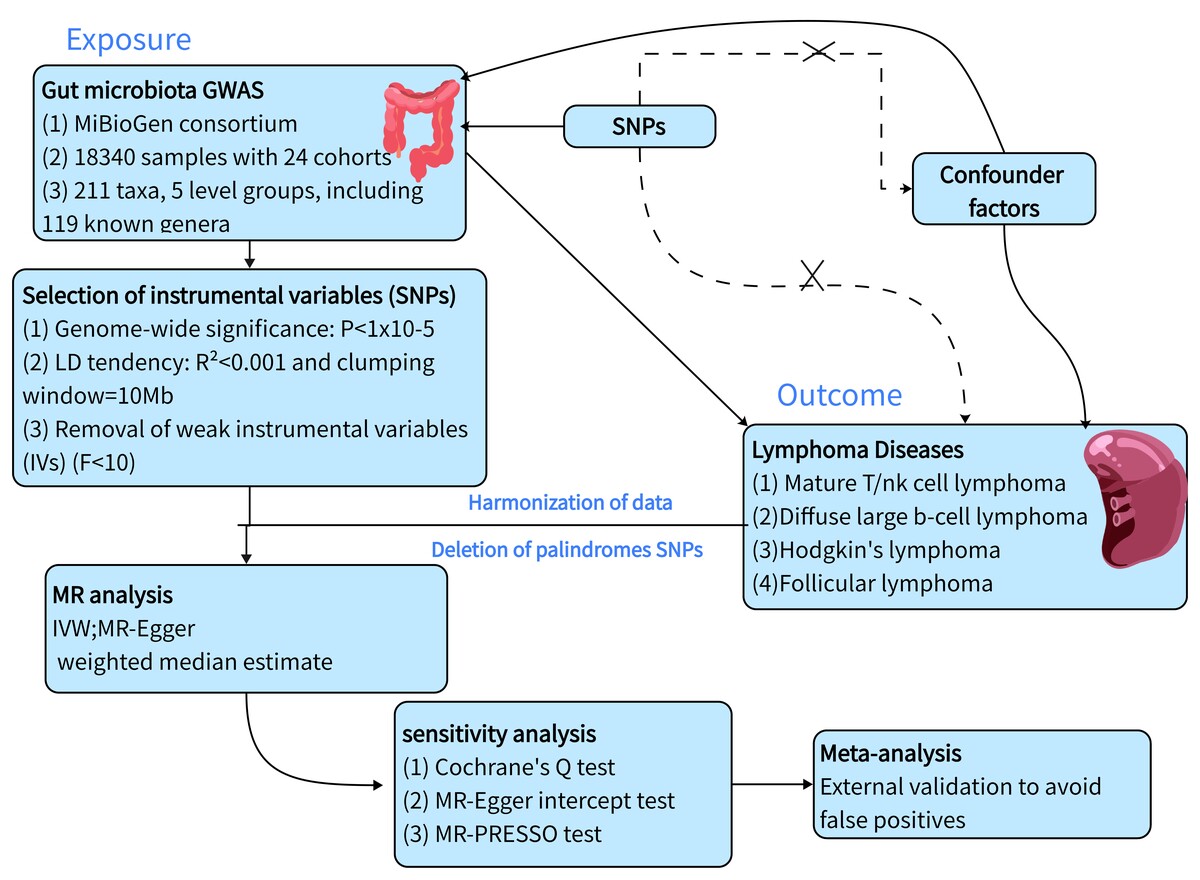
GWAS summary statistics that are available to the public were used in this study. There was no need for additional ethical approvals. The study flow chart is displayed in Figure 1.
We implemented rigorous quality control procedures to select the most robust IVs and ensure the validity and accuracy of our results. Initially, we selected SNPs strongly associated with GM, applying a significance threshold of p = 1 × 10–5. Secondly, an important step in MR analysis is to ensure that the effect of the SNP on exposure corresponds to the same allele as the effect on outcome. After matching the results, we removed the palindromic SNP. (Palindromic SNPs are SNPs with A/T or G/C alleles.) Thirdly, only independent SNPs were kept after clustering the SNPs in each bacterial classification unit. The linkage disequilibrium (LD) threshold was set to r2 < 0.01 and the clustering window size to 10,000 kb [25]. Furthermore, the influence of pleiotropy was eliminated by applying the MR pleiotropy residuals sum and outlier (MR-PRESSO) test, which was used to identify possible horizontal pleiotropy by removing outliers [26]. In addition, F statistics were computed in order to assess weak instrument bias F = (R2(n – 1 – k))/((1 – R2)k) < 10 was considered a weak IV, R2 is the proportion of genetic variation explaining exposure, N is the sample size of exposure, and k is the number of SNPs used for MR analysis. The weak instrumental variable was excluded from the subsequent analysis [12].
Statistical analysis
MR analysis
To investigate possible causal relationships between lymphoma and GM we performed MR analysis. The gut microbiome with multiple IVs was characterized using three prominent MR methods: the weighted median estimator (WME) [27], MREgger regression [28], and the inverse variance weighted (IVW) method [29]. If there was only a single IV, only the Wald ratio method was used to perform the analysis. The results for multiple IVs are mainly based on the IVW method, supplemented by the other two methods. This is because it has been observed that in some cases, the IVW method is more effective than other methods [30].
Sensitivity analysis
We tested for heterogeneity using Cochrane’s Q test. P < 0.05 was considered heterogeneous. The intercept of MR-Egger regression was used to assess the presence of horizontal pleiotropy in IVS, and MR-PRESSO was used to propose SNPs with pleiotropy. Finally, FDR correction was applied to reduce the possibility of false positives; PFDR < 0.05 was considered a significant result.
Validation based on external cohorts and meta-analysis
To further validate the reliability of the NMR analysis results, two large-scale external cohort studies were used to validate the NMR-positive results: the FINRISK study in Finland and the Lifeline study in the northern Netherlands. It is noteworthy that to enhance the robustness of the results, datasets that were at disparate levels of bacterial classification were excluded. In the absence of relevant cohorts in the datasets, a meta-analysis was not performed. The external cohort validation process was conducted using the identical parameters and procedures as those employed for the MR analysis described above. A meta-analysis was performed on the analyzed results to incorporate effect sizes. The I2 statistic was employed to assess heterogeneity in the meta-analysis. When I2 < 50% and p > 0.05, a fixed-effects model was utilized. Conversely, when I2 ≥ 50% and p ≤ 0.05, a random-effects model was employed.
R (version 4.3.2) packages that use TwoSampleMR, data.table, dplyr, doParallel, ggthemes, magrittr, p.adjust and readr were used for all statistical studies.
Results
Two-sample MR analysis of lymphoma
The F-statistics for all IVs were greater than 10, indicating that there is no weak IV bias in our analysis (Supplementary Table SI). MR analyses revealed 37 causal associations between genetically predicted GM and four lymphoma diseases. In Hodgkin’s lymphoma, we found 15 causal relationships. In follicular lymphoma disease, we found 11 causal relationships. In diffuse large B-cell lymphoma, we found 5 causal relationships. In mature T/NK-cell lymphomas, we found 6 causal relationships.
MR analysis showed that 6 GM were associated with an increase in the incidence of Hodgkin’s lymphoma and 9 were associated with a decrease in the incidence of Hodgkin’s lymphoma, with the more significant ones being genera. Butyricimonas (OR = 2.125, 95% CI = 1.173–3.849, p = 0.013) and order Bifidobacteriales (OR = 2.154, 95% CI = 1.117–3.962, p = 0.014) were significantly associated with increased risk of Hodgkin’s lymphoma. The genus Peptococcus (OR = 0.573, 95% CI = 0.390–0.840, p = 0.004) was associated with significantly decreased risk of Hodgkin’s lymphoma.
Three GM were associated with increased incidence of diffuse large B-cell lymphoma, and two GM were associated with decreased incidence of diffuse large B-cell lymphoma, the more significant of which were the Eubacterium genus group (OR = 0.236, 95% CI = 0.079–0.712, p = 0.01), which was negatively correlated, and the genus Terrisporobacter (OR = 3.607, 95% CI = 1.189–10.938, p = 0.023), which was positively correlated.
Four GM were associated with increased incidence of follicular lymphoma and seven were associated with decreased incidence of follicular lymphoma, the more significant of which were Eubacterium (OR = 0.450, 95% CI = 0.224–0.900, p = 0.024), which was associated with a reduced risk, and the phylum Proteobacteria (OR = 1.984, 95% CI = 1.063–3.704, p = 0.031), associated with elevated risk.
Two GM were associated with increased incidence of mature T/NK-cell lymphoma, and four GM were associated with decreased incidence of mature T/NK-cell lymphoma, the more significant of which were the genus Ruminococcaceae UCG003 (OR = 0.289, 95% CI = 0.102–0.821, p = 0.020), the genus LachnospiraceaeUCG001 (OR = 0.381, 95% CI = 0.161–0.901, p = 0.028), which were associated with a reduced risk of morbidity, and the order Lactobacillales (OR = 4.751, 95% CI = 1.802–12.530, p = 0.001), which was associated with an increased risk of morbidity. See Figure 2 for details.
Figure 2
MR results and its forest plot. Causal effects for gut microbiota on T/NK cell lymphoma; causal effects for gut microbiota on diffuse large B-cell lymphoma; causal effects for gut microbiota on follicular lymphoma; causal effects for gut microbiota on Hodgkin’s lymphoma
OR – odds radio, 95% CI – 95% confidence interval, IVW – inverse-variance weighted. P < 0.05 was considered statistically significant.
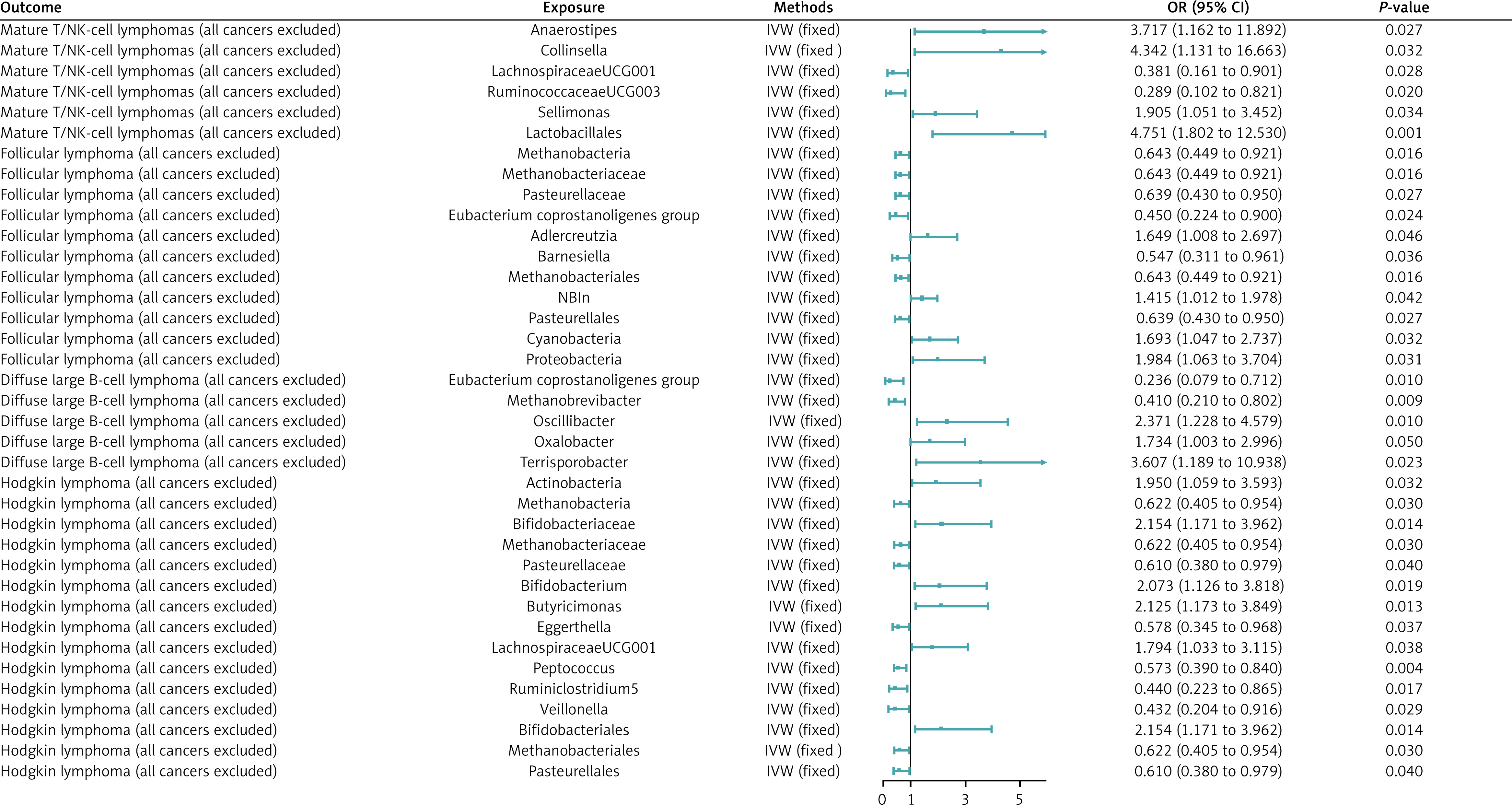
After FDR correction, Lactobacillales showed a significant causal association with Mature T/NK-cell lymphomas (OR = 4.751, 95% CI = 1.802–12.530, pFDR = 0.030). Detailed information is provided in Supplementary Table SII.
Sensitivity analyses of the 37 significant causal associations showed that neither MR-Egger nor MR-PRESSO analyses detected the presence of horizontal pleiotropy. In addition, there was no significant heterogeneity among the selected SNPs according to the Cochrane Q test (p > 0.05). Detailed information is provided in Table II.
Table II
Sensitivity analysis of the causal association between gut microbiota and lymphoma
Validation based on external cohorts and meta-analysis
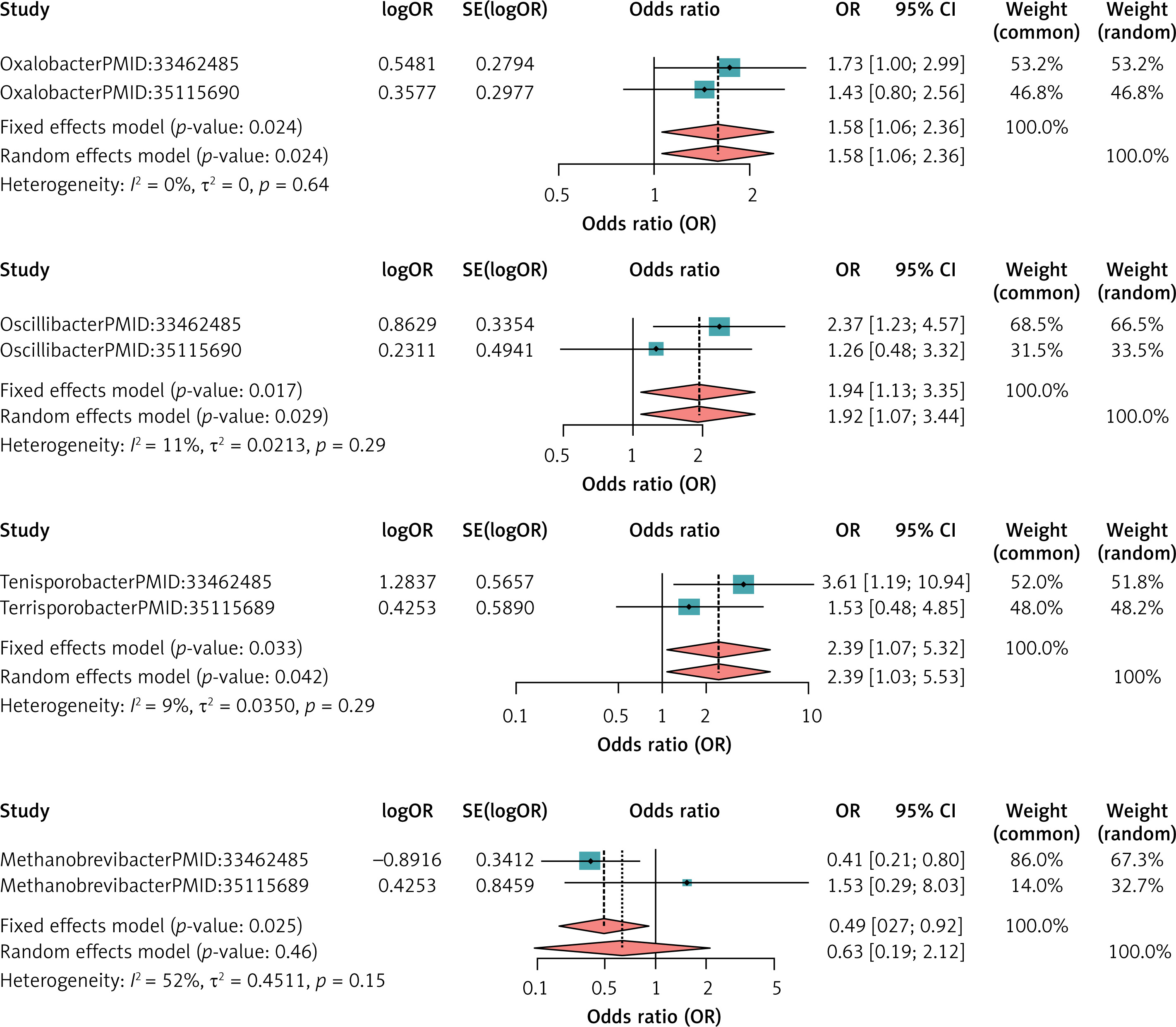
The dataset of 16 intestinal flora used to validate the results of the MRI analysis was obtained from the IEU Open GWAS project and is consistent with the present study at the level of intestinal flora classification to ensure good homogeneity. No meta-analysis was performed for the other flora as there was no corresponding dataset for them. For DLBCL, the heterogeneity test showed that no dataset with I2 > 50% and p < 0.05 existed, so all were meta-analyzed using a fixed-effects model. Meta-analysis results showed that Oxalobacter (MetaOR = 1.58, 95% CI = 1.06–2.36, p = 0.02), Oscillibacter (MetaOR = 1.94, 95% CI = 1.13–3.35, p = 0.02) and Terrisporobacter (MetaOR = 2.39, 95% CI = 1.07–5.32, p = 0.03) had a pathogenic effect on DLBCL. Methanobrevibacter (MetaOR = 0.49, 95% CI = 0.27–0.92, p = 0.03) was protective against DLBCL. See Figure 3 for details.
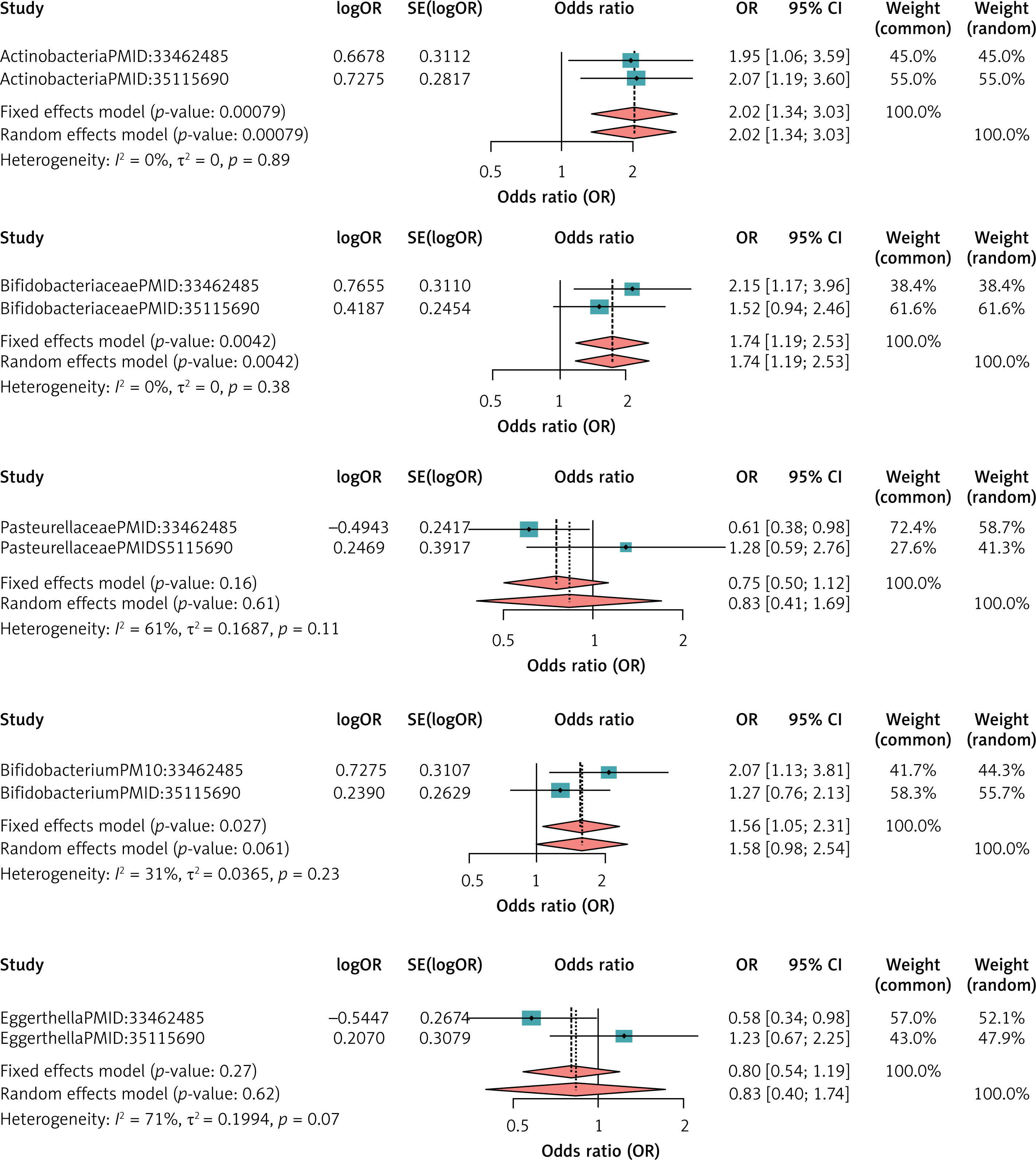
For HL, the heterogeneity test showed that there was no dataset with I2 > 50% and p < 0.05, so all were meta-analyzed using a fixed-effects model. The meta-analysis showed that Actinobacteria (MetaOR = 2.02, 95% CI = 1.34–3.03, p < 0.001), Bifidobacteriaceae (MetaOR = 1.74, 95% CI = 1.19–2.53, p = 0.004), Bifidobacterium (MetaOR = 1.56, 95% CI = 1.05–2.31, p = 0.02) had a pathogenic effect on HL and the other two bacterial groups showed no significance. See Figure 4 for details.
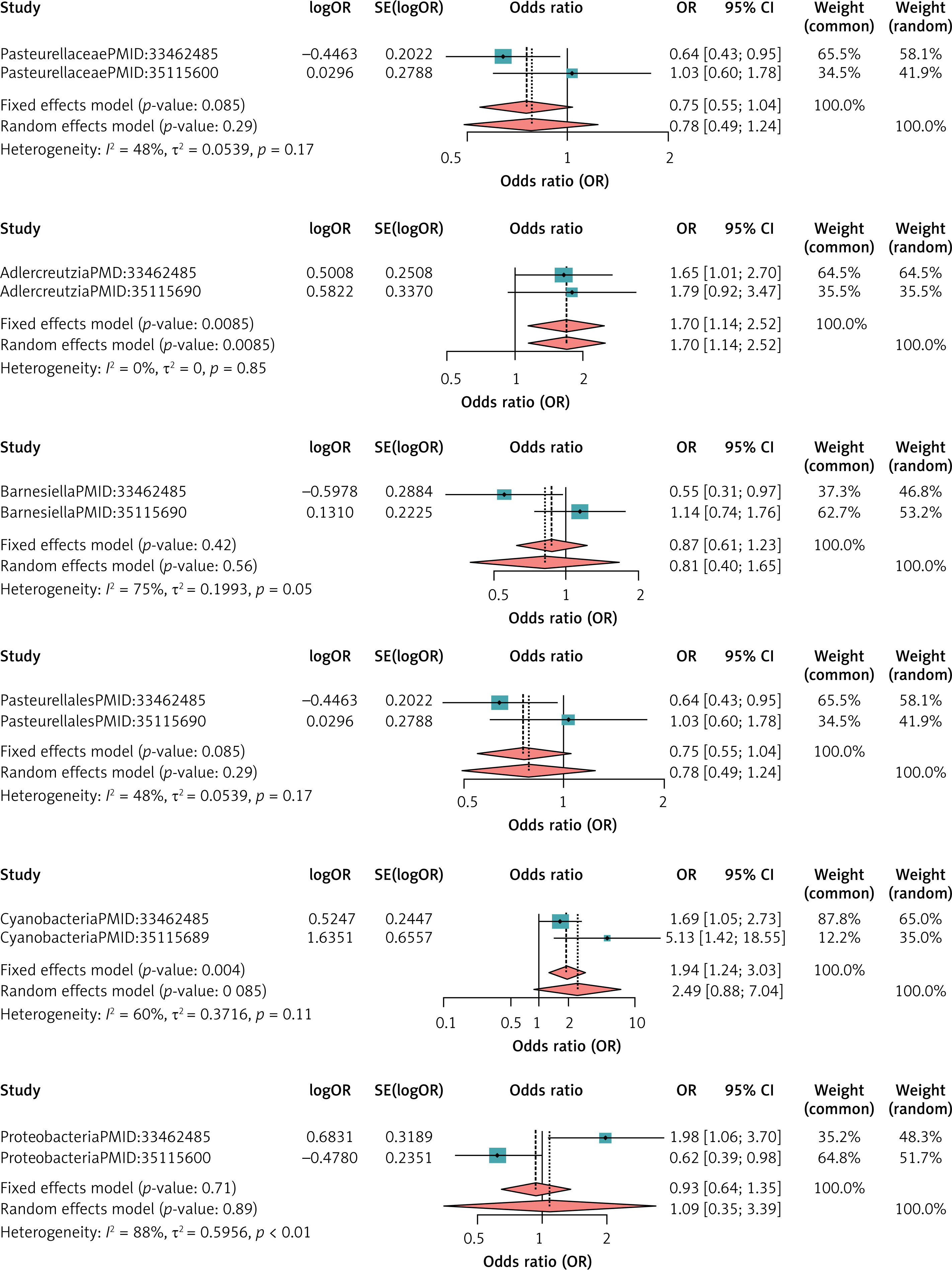
For FL, the heterogeneity test showed the presence of one colony with I2 > 50% and p < 0.05 in the dataset, so the meta-analysis was performed using the random effects model, and the other meta-analysis was performed using the fixed effects model, and the results of the meta-analysis showed the pathogenic effect of Adlercreutzia (MetaOR = 1.70, 95% CI = 1.14–2.52, p = 0.008), Cyanobacteria (MetaOR = 1.94, 95% CI = 1.24–3.03, p = 0.004) on FL, and the other results were not significant. See Figure 5 for details.
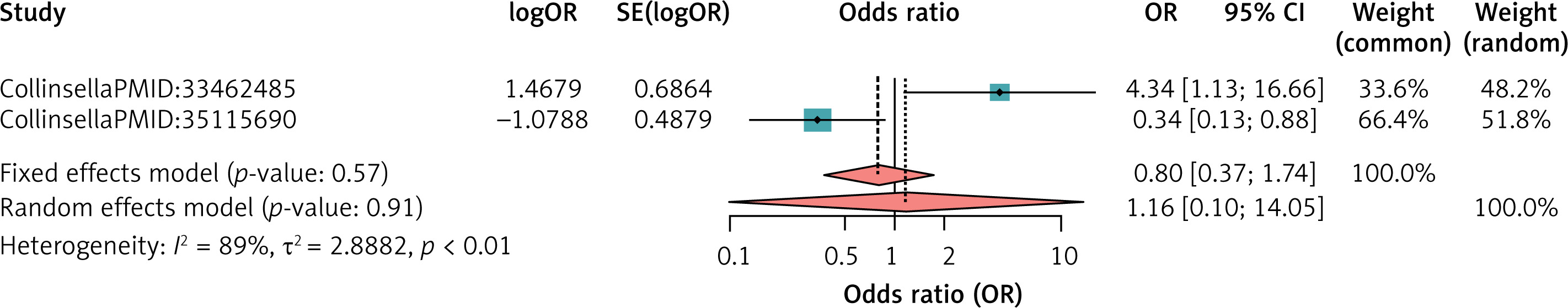
For NKTCL, the meta-analysis result was non-significant (Figure 6).
Discussion
In this study, MR analysis showed an association between gut microbiota and lymphoma. We used the extensive GWAS meta-analysis data on GM obtained from the MiBioGen consortium and lymphoma statistics published by the IEU to examine potential causal relationships. We identified 37 cases of GM with a significant causal association with lymphoma and performed a meta-analysis to obtain 9 cases of GM that remained significant.
Numerous studies have identified a possible link between the GM selected in our study and lymphoma. Our findings suggest that Eubacterium may act as a protective agent for diffuse large B-cell lymphoma and follicular lymphoma, which is consistent with previous reports [31]. A class of strictly anaerobic, Gram-positive, non-spore-forming bacteria known as the Eubacterium genus are frequently found in the human gut and are distinguished by their ability to produce butyrate [32]. Butyrate has strong anti-inflammatory properties, and early studies have shown that butyrate has anti-inflammatory properties through the interaction of G protein-coupled receptors GPR41 and GPR43 and strongly inhibits the release of the pro-inflammatory cytokine TNF from lamina propria mononuclear cells [33–35] in an inflammatory environment, as the pro-inflammatory cytokine TNF upregulates TLR4 in intestinal B cells. This makes B cells more sensitive to LPS [31]. TNF in combination with LPS may lead to lymphoma, while Eubacterium inhibits it, suggesting a correlation between Eubacterium and reduced risk of lymphoma [31].
The present study proposes that Terrisporobacter is a high-risk GM for the development of DLBCL and has the potential to be a specific marker or therapeutic target. Terrisporobacter is an anaerobic fungus that is frequently detected in postoperative patients suffering from comorbidities, such as abscesses and bloodstream infections [36], and is positively correlated with the risk of sepsis [37]. Furthermore, invasive fungal disease (IFD) represents a significant cause of morbidity and mortality in patients with hematologic malignancies.
Our study is the first to propose cyanobacteria as a high-risk GM for FL, and a study has suggested a possible carcinogenic pathway of cyanobacteria associated with colorectal cancer. Cyanobacteria produce a harmful secondary metabolite, microcystin-LR (MC-LR). MC-LR activates the PI3-K/AKT signaling pathway, leading to epithelial-mesenchymal transition, and regulates the expression of miR-221/PTEN and STAT3 signaling pathways, which promotes invasion and metastasis of CRC cells [38]. In addition, some epidemiologic studies have shown that long-term exposure to MC-LR increases the incidence of several cancers, including hepatocellular carcinoma [39] and CRC [40], and may lead to cancer progression [41]. Therefore, studies are needed to explore the mechanisms.
GM has also been studied as a non-invasive diagnostic and prognostic biomarker for natural killer/T-cell lymphoma [42]. There are few studies on Hodgkin’s lymphoma [15], and our study may provide some ideas for the impact of GM on Hodgkin’s lymphoma.
This study has several notable strengths and innovations. Firstly, we employed a novel approach combining MR analysis with meta-analysis of the combined results, which allowed us to investigate the causal relationship between GM and lymphoma in a way that has not been previously attempted. Secondly, our study is less susceptible to confounding factors and reverse causation than traditional observational studies, which allows us to infer the etiology of complex diseases with greater precision.
However, it is important to recognize the limitations of this study. First, ethnicity affects the results of gene-level studies. The genomics analysis data used in this study were primarily from European populations, which limits the possibility of generalizing the current findings to other ethnic groups. Larger studies are necessary to further corroborate these data. The control thresholds used in the selection of IVs were not stringent enough. Increasing the thresholds would help to identify more potentially valuable IVs. Second, this study did not externally validate all positive results, and there was a lack of data on some of the gut microbiota.
In conclusion, we performed MR analysis to determine the causal relationship between GM and four lymphoma diseases. In Hodgkin’s lymphoma, we found 15 causal relationships. In follicular lymphoma, we found 11 causal relationships. In diffuse large B-cell lymphoma, we found 5 causal relationships. In mature T/NK-cell lymphomas, six causal relationships were identified and some of the positive results were externally validated. Furthermore, for the first time, it was proposed that cyanobacteria represent a high-risk factor for FL, thereby providing new insights into the mechanisms of intestinal microbe-mediated lymphoma progression and diagnostic and therapeutic follow-up.